

A call to arms to save the genome
How protecting our genetic code can turn the tide against death


Introduction
Imagine your task is to maximize the amount of time a boulder takes to roll down a hill.

What are your options?
Apply some force of friction to slow down its acceleration downwards.
Push the boulder back uphill.
In the field of longevity, we are putting nearly all of our time, energy, and resources into option (2), via trying to reverse aging with rejuvenation techniques. Even worse, we almost always begin interventions once the boulder is already barreling at full speed, with every hallmark of aging manifesting itself in detrimental ways. Wouldn’t it be far more effective to act on the forces acting on the boulder at the top of the hill, and slow it down before it reaches high velocity?
TL;DR:
The most effective way to slow down the boulder is by addressing genome instability, ideally as early as possible.
There are clinically relevant areas for intervention where clinical trials could show promising results.
In a piece coming out in a few days (stay tuned!), I'll dig deeper into the science underlying these interventions and lay out a new framework for looking at the field of genome instability in the context of aging.
Table of Contents
The Idea
What is Aging, Anyway?
This is a contentious topic, with many differing opinions and viewpoints. Most published work on this tends to have a fairly lowest-common-denominator definition of aging that is agreeable to most but is also quite vague. Take this definition written by the Biomarkers of Aging Consortium:

The vagueness makes it challenging to concretely define whether or not an intervention “reverses aging” or not. Per the original analogy - is the intervention truly pushing the boulder back up the hill? Does Paxlovid reverse aging, as it is a treatment for COVID? COVID is, after all, a “consequence of life…. that leads to functional decline.” Do GLP-1 agonists reverse aging as they ameliorate age-related metabolic pathology (interesting debate on this here)? Does rapamycin reverse aging?
I’ll propose a different definition of aging. Let’s go back to the rolling boulder example from before, and imagine the boulder reaching the bottom of the hill as representing death.

The distance the boulder has traveled represents biological age - your risk of mortality and onset of age-associated diseases. This tracks nicely with the fact mortality increases exponentially with age (source):

Just as there is a constant force (gravity) forcing the boulder down the hill, so there is a constant force (the root cause(s) of aging) causing us to age. See the below graphic for clarity:

This is not an academic exercise - the implications of thinking about aging as a boulder rolling down a mountain are profound. Firstly, the ability to slow biological aging becomes exponentially more difficult later in life, so late-life interventions for individuals in their 80s are (literally) fighting an uphill battle. Secondly, claims of “reversing biological age” require a decrease in the risk of age-related diseases with time, which has yet to be demonstrated by any therapy in mice or humans. Thirdly, if the experiences of biological aging are exponential, then the rate of aging should have a linear (non-exponential) relationship with time and the root cause(s) of aging should be constant.
Lastly, and most importantly - by far the easiest way to increase the amount of time for the boulder to roll down the hill is not by trying to push it directly, but rather slowing its progression down the hill. This means tackling the acceleration of aging - its root causes. Thus, I call for improving genome stability.
But why genome instability?
(for the curious reader: the above model for aging is somewhat simplified, but it is remarkably similar to ideals posited by a few authors in the past [1] [2] [3])
Why Genome Instability?
If the effects of aging (such as mortality) have an exponential relationship with time, then the forces accelerating it (the root cause(s) of aging) should be constant across the lifespan, thus affecting a linear degradation in immediate downstream targets. Thus, a good way of hunting for the root cause(s) of aging is to find aging-associated hallmarks that change linearly with time. Here’s the boulder graphic again for clarity:

Linear relationships with age are relatively hard to come by. Protein aggregates, at least in yeast, show an exponential increase with age. Also, accumulation of many senescence markers (those related to age-associated nondividing & likely-damaged cells) in mice appears to dramatically increase towards the end of a mouse's lifespan. But what about something related to genome instability - such as somatic mutation frequency (from this paper)?
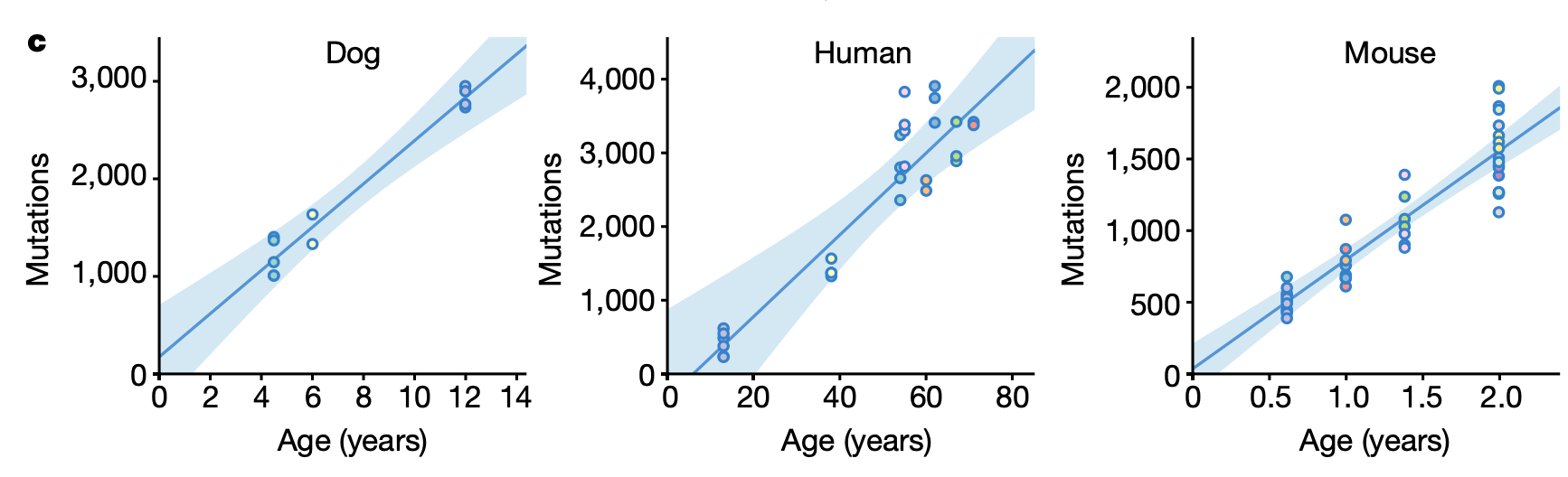
These look remarkably like straight lines. This is even more interesting when combined with the finding that average lifespan scales incredibly closely to mutation rate across species.

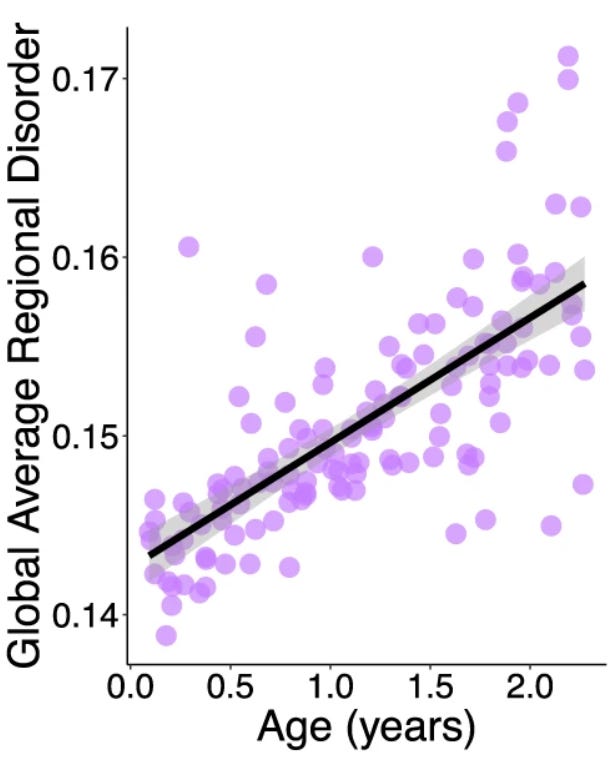
This effect isn’t limited to mutation rate, either. DNA methylation patterns in rats, dogs, and baboons (though probably not mice!) all shift with age in a linear fashion:
The evidence for the failure of genomic maintenance being a primary driver of aging is not limited to graphs with straight lines. When I initially entered the aging field, I was surprised to learn that every single human disease with a strong claim of being a form of accelerated aging has a mutation in a genome maintenance gene. Several years later, I have yet to find a satisfying reason for this to be the case without accepting DNA damage to be a significant cause of aging. Werner’s syndrome patients - those suffering from a mutation in a key cell replication and telomere maintenance gene - show multifold signs of aging, including graying hair, early onset diabetes, and cataracts. It’s worth noting that these patients do not demonstrate an accelerated form of every aging symptom (more on this in a soon-to-be-released piece), but they certainly do look much older:

If impairing genome stability shortens lifespan, does improving genome stability increase lifespan? Early data suggests this to be the case. A cohort of centenarians has been demonstrated to have a more effective variant of the critical epigenetic maintenance protein SIRT6. A genetic variant in FOXO3A, a gene encoding a protein involved in the DNA damage response (among many other functions), is one of the few that has been conclusively linked to human longevity. This also applies across the animal kingdom - DNA repair pathways are consistently more present in longer-lived species and quieter in those that die sooner.
Where are We Now?
The microenvironment of individuals picking up the mantle of fixing genome stability in the context of aging could best be described as a “niche.” The field is, more than anything, currently focused on understanding the mechanistic drivers relating genome stability to aging. Nevertheless, the first glimmers of an effort to intervene in the root cause of aging can be found, both in academia and industry. To contextualize these endeavors in increasing levels of difficulty, I have split them into four buckets of approaches that improve genome stability by:
Preventing DNA damage and instability.
Inhibiting factors that prevent DNA repair and stabilization.
Activating processes that reinforce DNA repair or organization.
(Next-generation) Harnessing gene editing or capabilities from other animals to improve genomic maintenance.
Charting the Course
Tier 1: Prevent DNA Damage

The most elegant way to prevent genome instability is to stop damage from happening in the first place. Unfortunately, this is a challenging proposition. DNA damage can come from nearly anywhere, including the sun and reactive oxygen species (ROS) derived from normal metabolism. Equally unfortunately, UV light is required for production of vitamin D, and ROS are used as part of healthy signaling processes throughout different tissues in the human body. High intake of antioxidants, the most well-known compounds thought to prevent DNA damage, is not associated with lower all-cause mortality. Cells have evolved incredibly sophisticated mechanisms for managing potential DNA damaging agents in a manner that makes modulation difficult.
Opportunities for interventions are still possible, however, particularly if implemented in a way that works cooperatively with cellular machinery. Taurine supplementation shows improvement in lifespan in mice while decreasing multiple types of DNA damage. Vitamin D may be protective against UV-induced DNA damage via an interesting mechanism involving preservation of expression of long genes. Many other supplements and natural products have been suggested to have effects on DNA damage prevention. Unfortunately, the literature on them remains murky at best - the effect sizes tend to be small, the compounds themselves prone to false positive readouts, and the dosing required to see an effect is frequently extremely high. However, the promise here is high - If a compound was discovered that could reliably block a type of DNA damage without considerable downside effects, it would be the holy grail of the genome stability field. So far, however, the results have been largely disappointing.
Tier 2: Repress Genome Destabilizers

Repressing a genetic pathway is the bread-and-butter of the biopharmaceutical industry. The best way to develop a drug is to find a chemical that blocks a deleterious pathway. If that’s not possible, find a chemical that blocks an inhibitor of a beneficial pathway. Unfortunately, this approach finds limited traction in the genome instability world. There are, generally speaking, precious few reasons for a cell to inhibit its own gene maintenance pathways unless it is actively dividing or apoptosing (in which case, it is generally not a good idea to meddle). Still, there are a few possibilities here.
Some intriguing possibilities for blocking repressors of genes that improve genome stability could be found in germline cells, as they are generally thought to be more effective at genomic maintenance. The Schumacher lab demonstrated that the inhibition of one such repressor, DREAM, led to an increase in DNA repair capacity. Unfortunately, as in many such cases, the DREAM complex also has key roles in cell cycle progression. Though this could prove problematic in dividing cells, such an inhibition in permanently non-dividing cells (such as neurons) could have far fewer drawbacks. There is an exciting possibility that other repressors such as DREAM remain undiscovered, possibly including those with a lesser degree of involvement in other critical cell processes.
Transposons, segments of DNA that replicate themselves and insert themselves in problematic places in the genome, are a known destabilizer of genetic architecture. Given that they consist of approximately 20% of the human genome, cells are forced to expend significant energy to repress them. Nucleoside reverse transcriptase inhibitors (NRTIs), drugs first deployed against HIV, have been shown to decrease age-associated inflammation by specifically inhibiting a type of transposon. This has, unsurprisingly, caught the attention of commercial actors. Transposon Therapeutics recently released positive Phase II data in a study focusing on progressive supranuclear palsy. Very recently, Altos Labs (a secretive longevity company funded by Jeff Bezos) has also been reported to be working on an oligonucleotide approach to treat accelerated aging disorders by inhibiting transposons. These are exciting developments, though it is worth noting the connection between transposon activation and human aging is suboptimal.
Tier 3: Activate Genome Stabilizers

The most intuitive way to improve genome stability is via upregulating or activating the proteins playing a role in maintaining it. Unfortunately, this is not so simple. The genome is closely regulated, and there are contexts in which optimal maintenance is not ideal. Persistent activation of repair responses also sends a signal to the cell that something is terribly wrong - and the cell can react by activating persistent inflammatory signaling or apoptosing. Even if a safe target were found, a small molecule activator for it may not exist. The search for such a molecule would be especially difficult as those that could be utilized in the context of genome maintenance will need to be both safe and specific.
This is not to say that a small molecule activator improving genome maintenance would be impossible to find. Fairly recently, an activator of an enzyme involved in repairing oxidative damage was found to function via an unconventional mechanism. This is particularly exciting, as the enzyme is responsible for directly repairing DNA damage as opposed to being a master regulator of a complicated network. A number of compounds, including resveratrol, have also controversially been named as sirtuin activators capable of extending lifespan. Some evidence, too, suggests that NAD+ precursors play a role in boosting DNA repair capabilities, though the mechanism is not clear. Also, farrerol could be an activator of oxidative stress resistance pathways. Overall, our current arsenal of genome stabilizers remains extremely small. This is not only due to the complexity of the underlying biochemistry, but is also the result of a lack of appreciation for the potential of such molecules.
Tier 4: Beyond Small Molecules

Small molecules are a fairly indirect method for improving genome stability or preventing damage. What if we could simply directly increase the expression of maintenance mechanisms in humans? Alternatively, what if we could pull mechanisms from other animals and incorporate them into ourselves? Numerous companies are taking on these approaches already.
Genflow Biosciences is rapidly seeking to transfer the centenarian SIRT6 variant into the rest of us, by first seeking to treat Werner’s Syndrome and NASH. Matter Bio is finding ambitious ways to reverse the mutations that have already occurred, in some way breaking the conventional wisdom that improving genome stability will only slow the rate of aging. Peel Therapeutics is seeking to deliver an elephant copy of a key “guardian of the genome” gene.
Many of these approaches come with caveats:
Most applications for improving genome instability are relevant in the context of preventing a disease rather than curing one. In the grand scheme, the hope is to deploy them across all healthy adults. This is not quite compatible with gene delivery approaches, which do not currently have a safety profile that makes them compatible with use in healthy adults. As a result, the use of these techniques (for now) may only be in patients with a devastating disease.
The durability and targeting of gene delivery approaches is still a work in progress, limiting administration to certain tissues or organs. Some areas of the body (such as the brain) where improved genome stability would be the most useful are also the most difficult to deliver genes to.
The underlying molecular mechanisms and pathways of many genes involved in genomic maintenance are unclear, and therefore the targeted disease indications may be challenging to identify.
None of these technical challenges are insurmountable, and incredibly ambitious approaches will need to be piloted in order for improvements in human maximum lifespan to occur.
Each tier comes with its trade-offs. Preventing DNA damage seems like the most intuitive and easiest approach, and using gene therapies to up-regulate repair is the most challenging. Yet from an evolutionary point of view, it is likely that our bodies have already evolved advanced mechanisms to deal with endogenous sources of damage, and it is only through the more radical approaches that we can push our defenses past their limits. The best-of-both-worlds approach may be to do something specific and context-dependent. For example, many Tier 3 approaches (those increasing the expression of genome stabilizing genes) lead to permanent cycle arrest, but this could be used in a cell type that is already post-mitotic. Alternatively, a Tier 1 approach may prove to be beneficial in the context of ameliorating rarer types of damage that the cell has not evolved adequate defenses against.
Regardless of the intervention, it will need to be paired with a disease to correct (at least in the short-to-medium term) for FDA clinical trials. I have listed a few promising areas below.
Places to Intervene
Prevent Cancer

As cancer is the second leading cause of death in the United States and is linked very tightly to failure of DNA repair, preventing it is a promising avenue through which to tackle improvements in genome stability more generally. Cancer prevention has been done before - tamoxifen can help prevent breast cancer and aspirin could help lower the risk of colorectal cancer. Nicotinamide, a molecule familiar to those in the aging space, has been shown to decrease skin cancer incidence in individuals who had previously been diagnosed with melanoma. The idea of using a drug to prevent cancer is extremely appealing, particularly as the potential patient population immediately includes everyone. There are a few caveats and challenges, however:
Not every genome maintenance pathway relevant to cancer is necessarily tied to aging.
Clinical trials for cancer prevention tend to be difficult and expensive. Even in the elderly, the annual incidence of cancer is quite rare, meaning trials that seek to prevent cancer in the healthy adult population will need to enroll many thousands of participants.
If the drug is going to be prescribed to everyone, it has to be extremely safe. This is generally true for any longevity drug but becomes acutely important in the context of long-term administration to prevent a possible future case of cancer rather than treating an acute disease.
Nevertheless, the possibility of being able to tackle both aging and cancer in one fell swoop is incredibly enticing.
Delay Menopause
The age of menopause is closely tied to women’s overall health (more on this in a wonderful post authored by Carol Magalhaes here!). There is intriguing evidence suggesting that improving DNA repair could be a direct method by which to delay the onset of menopause by prolonging the longevity of oocytes. In Genome-Wide Association studies (GWAS) investigating genetic associations with the age of onset of menopause, DNA repair genes frequently emerge as top hits. Variants in DNA regions near EXO1, one of the genes that emerged as a top hit, have also been shown to be linked to cognitive aging in a Taiwanese population and associated with prolonged life expectancy in centenarians.
The mechanism here is unclear - it is possible that DNA repair keeps oocytes alive for longer and thereby directly delays menopause by maintaining the ovarian reserve, or it could have other functions in maintaining other ovarian cells or those involved in neuroendocrine signaling. Regardless, this is a promising avenue through which to significantly improve women’s health.
Slow Neurocognitive Disorders

Though oxidative damage to DNA occurs widely throughout the body, the brain appears to be uniquely sensitive. The reasons for this are likely multifaceted but may be linked to the inability of neurons to divide and thus dilute out damage, or caused by the high metabolic load in the brain. Deficiencies in oxidative damage repair, detection, or prevention are linked to ALS, Ataxia-Telangiectasia, and Cockayne syndrome. Each of these three diseases has a pronounced neurodegenerative component, suggesting a central role of oxidative damage in destabilizing genome instability in neurons. Less causally, oxidative stress has also been implicated in the pathogenesis of Alzheimer’s disease (even at very early stages!) and Parkinson’s disease. Depletion of BRCA1, a protein responsible for DNA repair, has been shown to occur in Alzheimer’s disease brains with possible downstream effects on memory and cognitive function. Similarly, XRCC1 variants have been coupled with an increased risk of Alzheimer’s in a Polish cohort.
Diseases aside, cognitive capabilities are one of the first functions to decrease in humans with age. In the brain, DNA damage wrecks havoc long before we start to acutely notice its effects. If we can find ways to improve our resilience, ideally as early as possible, we can both slow the incidence of disease and maintain our ability to think.
Stabilize the Brain Post-Trauma

Given that the brain is sensitive to oxidative damage, it will not surprise you to learn that events such as a stroke or seizure cause enormous amounts of DNA damage. Curiously, there’s evidence to suggest that failure to resolve DNA damage could even be causal for epilepsy, as over-activation of DNA damage sensors can lead to seizures in mice. Rare mutations in a single-strand DNA repair gene in humans manifest primarily as seizures and neurocognitive impairment. Debate exists around whether the true cause of neuronal cell death in the case of strokes and seizures is the DNA damage itself or a hyper-activated response to it. If you knock out a key DNA damage detector (PARP1) in mouse brains, they become much more resistant to neuronal damage induced by a stroke. The mechanism for this effect is also unclear, but could either be linked to direct cell death mechanisms or through DNA damage sensors burning through key energy metabolites that could be more effectively spent on repairing other forms of damage. In any case, if repair could be made more efficient then the amount of energy consumed for DNA damage signaling could be reduced, leading to neuronal protection.
Hamper Fibrotic Disorders

In the public eye, telomeres have been long connected to aging. If you work in the longevity field and mention that you are working on aging, a very common response is “oh, so like telomeres and stuff?” Unfortunately, the link between telomeres and aging in humans has proven to be fairly elusive. The connection is strong, however, in some diseases including pulmonary fibrosis. The gene encoding TERC, a component of telomerase (protein that extends telomeres), leads to inheritable pulmonary fibrosis when mutated. In mice, over-expressing telomerase leads to lung regeneration in models of fibrosis.
The connection between genome instability and fibrotic diseases extends beyond telomeres. As an example, over-expression of a gene that promotes the formation of a key DNA repair complex leads to disease improvement. In kidneys, over-expression of a protein responsible for the repair of oxidative DNA damage in the context of fibrosis led to an improvement in outcomes and, surprisingly, a positive effect on inflammation. Critically, many of these treatments were administered after the damage already occurred, suggesting that improving DNA repair could be used not solely in a preventative context.
Venturing into the Unknown

Saving the genome will not be an easy task. The number of drugs we currently have on hand to address it is extremely limited. Doing it wrong means driving up a pathological response that brings with it inflammation and cell death. Nevertheless, the promise is too great to ignore. There is definitive evidence for the central role genome instability plays in our aging process. By tackling it, we have the potential to address healthspan and lifespan in one swoop. Instead of hopelessly seeking to push the boulder of aging back up the hill once it has already reached a critical velocity in some tragic emulation of Sisyphus, we can instead directly affect the exponential process hurling us towards death. Slowing down the rate of aging by maintaining the genome even 10% better could bring each of us decades of life. It is up to us to take up this mantle and protect our genomes to live longer, healthier lives.
Acknowledgments
This piece would not have been possible without months of conversations, editing, revising, and thinking. Discussions with the following experts informed several critical components of this piece:
Dr. Jan Vijg
Dr. Laura Niedernhofer
Dr. Alex Cagan
Dr. Jan Hoeijmakers
Dr. Richard Frock
Dr. Ashby Morrison
Dr. Matt Yousefzadeh
Dr. Karim Mekhail
The age1 team (Alex Colville, Kat Kajderowicz, Carol Magalhaes, Maggie Li, and Alex Kesin) provided helpful edits, insights, and the fire needed to push this piece out into the world.
Conversations with genome stability in aging enthusiasts (Ruxandra Teslo, Zane Koch, Ronnie Cutler, Stacy Li, and many others) provided joy in writing this piece through inspiring creativity, brainstorming, and passion.
Lastly, a thank you to the Buck Institute genome stability fan club (Edward Anderton, Carlos Galicia, Sierra Lore, and Brendan Hughes). They are instrumental in creating a warm intellectual environment that combines a childlike sense of wonder with the zeal necessary to desire to make a difference in the world.
Appendix 1: Challenges
Models
The most common studies for DNA repair tend to expose cells to extremely high doses of a genotoxic agent (e.g. X-rays, UV, IR, cisplatin, doxorubicin) to induce high levels of DNA damage. The ability of a cell to resist this damage is representative of their DNA repair capacity, and screens that improve a cell’s resistance are noted as candidates for improving genome stability. Unfortunately, it is unclear if this is relevant for aging, given that different genotoxic agents cause unique types of genomic instability. Cells have diverse mechanisms to deal with threats to their encoding information, and it is unclear which types of defenses are relevant to aging. Even the applicability of cell models of accelerated aging disorders in humans is a topic of ongoing discussion. Worse still, it is unclear to what extent the same genomic maintenance pathways that fail in mice, worms, or yeast are translatable to humans. Werner’s syndrome models in mice fail to recapitulate the same accelerated aging phenotype as seen in humans, while Hutchinson-Gilford Progeria Syndrome models appear much more similar. Animal and cell models will be essential for understanding genome instability in humans, but we will need to deploy innovative techniques to ensure translatability.
Studying longevity in humans is challenging. Interventional lifespan studies are nearly impossible, as is nearly any type of aging-related exploratory investigation. Lifespan studies in mice are better, but take years. Worms, flies, and other species are faster - but with distance to humans come hints that findings may not translate. The evidence that core longevity mechanisms are conserved is enticing, highlighted by primordial pathways such as IGF signaling and addressed by interventions such as caloric restriction, but by no means is it clear that this is true for the entire causational aging spectrum. The Intervention Testing Program, a standardized protocol for testing potential longevity interventions in mice, has been a graveyard for many enticing approaches first discovered in lower organisms. Worse still, we will likely never know the potential approaches that would work in humans yet fail in mice.
The challenge of translating animal data into human clinical trials is only more difficult for work in genome stability. Effect sizes are unlikely to be massive immediately. Optimal bands of effectiveness will be narrow at first until we reach a greater holistic understanding of how the cell maintains its genetic information. To succeed, we will need far better models. Genome stability will need to be simulated in a translatable context, and perturbed in manners that can recapitulate the way a cell naturally responds to the aging process. A significant amount of theoretical work will need to be undertaken to decouple the phenomena of cancer from aging. Creating the framework that could lead to successful interventions in humans is the first step in turbocharging the field.
Readouts
In many ways, the genome stability field is still in its infancy with regards to capacity for scaling. Many labs have their own in-house assays for different DNA repair processes, and few of these have been cross-verified by other groups. The most widely-used assay, the comet assay (so called due to measuring a DNA smear that looks somewhat like the tail of a comet) was developed 40 years ago, is low-throughput, and only measures a small number of types of DNA damage. The gold standard approaches, including those based on mass spectrometry, tend to be very expensive. There are researchers currently focusing on single assays that can measure many different types of DNA repair simultaneously, and the field is increasingly working towards cross-validating different techniques. Nevertheless, there would be an enormous benefit from having dedicated, inexpensive, high-throughput, and trusted ways to measure different types of DNA repair and stability.
Clinical Trials
Fixing genome instability will solve, largely, a problem with the rate of aging. Optimistically, upregulation of relevant enzymes may fix DNA lesions that have accumulated with time, or alleviate pressure on systems that have not been able to keep up with damage occurrence. Nevertheless, as improving genome maintenance is not likely to be immediately rejuvenative, the benefits in the short-term will be less obvious than any possible therapy that reverses aging, even though the likelihood of success in the long-term may be greater. Additionally, effectiveness will be greater the earlier the intervention has begun - it is entirely possible that as a person ages, other downstream consequences take on a dominant role in age-related decline. This poses challenges for clinical trial readouts - even the XPRIZE Healthspan competition requires a gain in function, which is theoretically impossible if the rate of aging is slowed rather than reversed. The benefit of going after the slowing of aging anyway, of course, is that the probability of meaningfully increasing human lifespan by improving genome stability is much higher, even if the results are not seen immediately.
Appendix 2: Opportunities
Decades of Cancer Research
Ironically, prime candidates for improving genome stability may be found in lists of the genes mutated in chemotherapy-resistant cancers. Cancers, in some ways, become gain-of-function screens for genome stability in their own right when subjected to genotoxic agents. Some tumors improve their pathways to cut out DNA lesions to evade the effects of cisplatin. Cancers can “learn” to improve double-strand break repair to survive radiotherapy.
It will take time and effort to discover which pathways are key to slowing genomic attrition in aging. Peto’s paradox (the observation that larger animals do not necessarily get more cancer) means that animals that grow larger need to solve the problem of getting more cancer, and not all of these solutions slow down the rate of aging. Cancer could, nevertheless, be a promising avenue to identify which genes are most necessary for the successful repair of defined types of DNA stress. These “limiting” genes can then be screened for their impact on lifespan and healthspan.
Screening and Artificial Intelligence

With hundreds of proteins carefully titered by the cell for a given context, the likelihood of discovering the key interventions in genome stability by chance alone is extremely unlikely. Finding a way to scale will be fundamental to accelerating the pace of discovery. But this is much easier said than done - in screening, the devil is always in the details. A survival screen in the presence of a genotoxic agent may be a good place to start (and has been done by several groups), but enrichment may be found for genes involved in genotoxic agent efflux or senescence rather than core DNA maintenance machinery. It is also difficult to know which genotoxic agent best mimics the effect of aging.
Given good models and screening approaches, artificial intelligence may be an inflection point creator for the field. The recent discovery of a more effective SIRT6 variant in centenarians would almost certainly never have been found by conventional screening, no matter how well-resourced. Evolution was ultimately the generator of this improvement, with the improved variant ironically a product of failed DNA repair. Artificial intelligence, even at this early stage, is able to generate improved versions of currently existing human proteins. These approaches, especially if combined with a built-in computational understanding of the aging process in humans, could rapidly accelerate our progress in this domain.
Appendix 3: Labs Currently Working on Genome Instability in the Context of Aging
This is a table of a small subset of current labs that are tackling genome instability in the context of aging. This list will be updated & curated frequently

Appendix 4: Companies Addressing Genome Instability
This is a table of current companies that are tackling genome instability in the context of aging. It will be updated frequently

Appendix 5: Interventions Improving Genome Stability
This is a table of current small molecules that have evidence of improving genome stability and improving healthspan and/or lifespan.
