

Founder’s Playbook: From Target to Development Strategy
The five decisions between a validated target and a clinical program


Finally, the last piece of the series…...
One has to acknowledge that different types of targets (e.g. an extracellular enzyme vs. an intracellular protein vs. a gene therapy target) and different disease contexts demand different playbooks. We connect all the prior analyses to practical plans, ensuring that our strategy to drug the target and prove its value is as sound as the biology itself. I have listed the key considerations below for founders with identified targets looking to initiate the development phase.
1. Timeline to conviction
The central insight here is that target biology sets a floor on how fast you can reach human proof-of-concept, and everything else (trial design, financing, endpoints) builds from that constraint.
Fast-readout symptomatic targets (hours to weeks)
Vertex Pharmaceuticals / Suzetrigine (NaV1.8 inhibitor, acute pain) → clean example of genetically validated target enabling rapid conviction. NaV1.8 loss-of-function mutations cause pain insensitivity in humans. Vertex designed Phase 2 around post-surgical pain models (abdominoplasty, bunionectomy) where the primary endpoint - pain reduction at 48 hours - gives a binary signal in under a week per patient. Published in NEJM August 2023, positive Phase 3 readout January 2024, FDA approval January 2025 as Journavx. From Phase 2 publication to approval in roughly two years, making it one of the fastest first-in-class timelines in recent memory.
Disease-modifying targets requiring years of patience
Denali Therapeutics / BIIB122 (LRRK2 inhibitor, Parkinson’s disease). The Phase 2b LUMA study enrolled ~640 early-PD patients; clinical readout is expected in 2026 (roughly 6+ years after Phase 1 initiation). Denali’s strategy for surviving this timeline: (1) a $560M upfront + $465M equity Biogen partnership to share Phase 2b costs, (2) CSF and urinary LRRK2 phosphorylation biomarkers to demonstrate intermediate target engagement, (3) a diversified pipeline with nearer-term assets (tividenofusp alfa BLA submitted 2025) to provide interim milestones, and (4) $1.28B cash runway extending into 2028 plus a $275M Royalty Pharma deal. The $422.8M net loss in FY2024 shows the capital intensity of disease-modification programs.
Novartis / Pelacarsen (antisense, Lp(a) lowering for CV events). This is the extreme end: an event-driven cardiovascular outcomes trial where the timeline is partly beyond management’s control, it depends on MACE events accumulating in the study population. Results originally expected 2025, now pushed to 2026. Only a large-pharma balance sheet can absorb this kind of timeline uncertainty.
GLP-1 agonists in Parkinson’s (lixisenatide). Phase 2 results in NEJM (April 2024) showed lixisenatide slowed motor disability progression at 12 months. But confirming disease modification in neurodegeneration (distinguishing symptomatic from disease-modifying effects) requires years of follow-up. Multiple parallel GLP-1 PD trials (exenatide, NLY01, PT320) are now running, pushing the field toward biomarker-intermediate strategies using alpha-synuclein seeding assays and NfL levels.
Clever trial designs compress the timeline
Spyre Therapeutics - 9 proof-of-concept readouts in 2 years. Spyre is running two innovative designs simultaneously: SKYLINE-UC, a Phase 2 platform trial in ulcerative colitis testing three next-gen monotherapies (anti-α4β7, anti-TL1A, anti-IL-23) AND their combinations under a single protocol; and SKYWAY-RD, a basket trial evaluating anti-TL1A across three rheumatic diseases (RA, PsA, axSpA). With $757M in cash and runway into H2 2028, Spyre’s entire corporate strategy is organized around maximizing POC readouts per dollar.
Abivax / Obefazimod (UC, 8-week induction). Even though UC is a chronic inflammatory condition, Abivax designed an 8-week induction endpoint (Modified Mayo Score) that delivered Phase 2b proof-of-concept in months rather than years. Long-term extension then built progressive conviction (52.5% clinical remission at 96 weeks). Phase 3 ABTECT-1 and ABTECT-2 hit primary endpoints in July 2025; stock surged >500%. NDA submission planned H2 2026.
Regulatory pathway stacking as strategy
Breakthrough Therapy Designation has been granted to 634 of 1,622 requests (39%), and it reduces clinical development time by roughly 30%. Parexel analysis found that 52% of orphan drugs received BTD, and 60% of approved orphan BTD drugs stacked 2+ expedited programs. The most aggressive example: Krazati (adagrasib) achieved a 4.1-year development timeline (versus the 10.1-year average) by stacking accelerated approval + BTD + Fast Track + Priority Review.
Recent notable BTDs: Lilly’s sofetabart mipitecan (FRα ADC, 50% ORR across all FRα expression levels, January 2026); Cogent’s bezuclastinib (systemic mastocytosis, October 2025); Sanofi’s tolebrutinib (non-relapsing secondary progressive MS, December 2024).
2. Biomarker plan
The FDA-NIH BEST (Biomarkers, EndpointS, and other Tools) glossary defines a biomarker as “a defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions.” Molecular, histologic, radiographic, or physiologic characteristics are types of biomarkers. Important: biomarker is NOT an assessment of how a patient feels, functions, or survives. This distinction separates biomarkers from clinical outcomes, and confusing the two has destroyed billions in value.
The FDA recognizes seven biomarker categories: susceptibility/risk, diagnostic, monitoring, prognostic, predictive, pharmacodynamic/response, and safety. For drug development strategy, founders need to think about biomarkers in a chain (from molecule to patient) where each link answers a different question.
Target engagement biomarkers that informed dose selection
Eli Lilly / Donanemab, the “treat-to-target” paradigm. Donanemab used amyloid PET as a treatment-stopping biomarker: when patients achieved amyloid-negative status (<24.1 centiloids), dosing was stopped. 67.8% of patients reached this threshold by 76 weeks. In this case, a target engagement biomarker dictates not just dose but treatment duration.
Mechanism biomarkers versus disease biomarkers: the critical distinction
The anti-amyloid Alzheimer’s programs provide a clear illustration of this hierarchy:
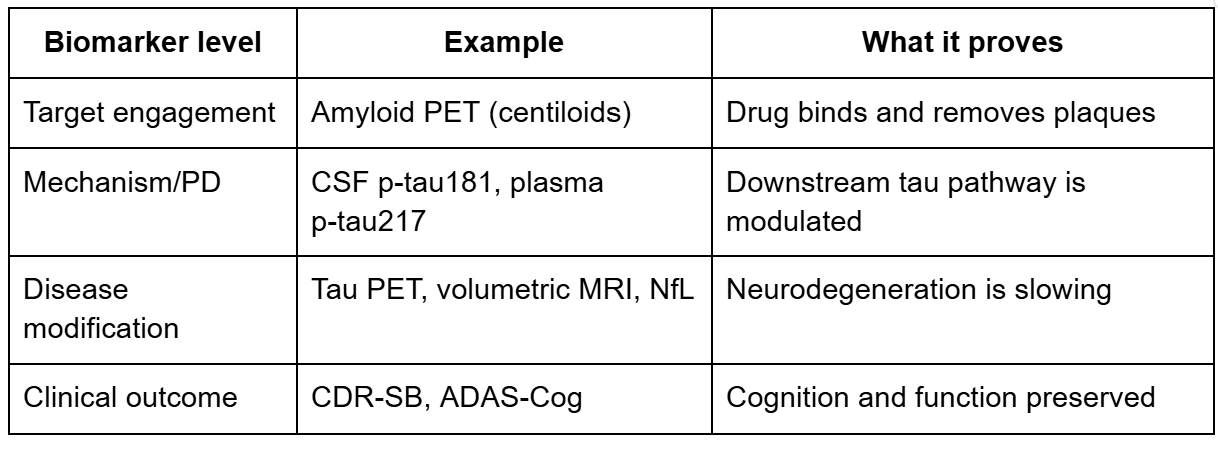
The critical lesson here: lecanemab produced large amyloid reductions (target engagement) AND CSF p-tau181 reductions (mechanism), but volumetric MRI paradoxically showed greater brain volume loss in the treatment group. Mechanism confirmed but disease biomarker gave mixed signals.
Alector / Latozinemab (anti-sortilin, FTD). The Phase 2 INFRONT-3 trial (2025) showed a “significant effect” on progranulin levels, the mechanism biomarker moved in the right direction. But MRI, fluid biomarkers of neurodegeneration, and clinical decline showed no apparent effect. Alector laid off ~50% of workforce and abandoned the program entirely. The drug clearly hit its target but failed to modify the disease. Mechanism engagement ≠ disease modification.
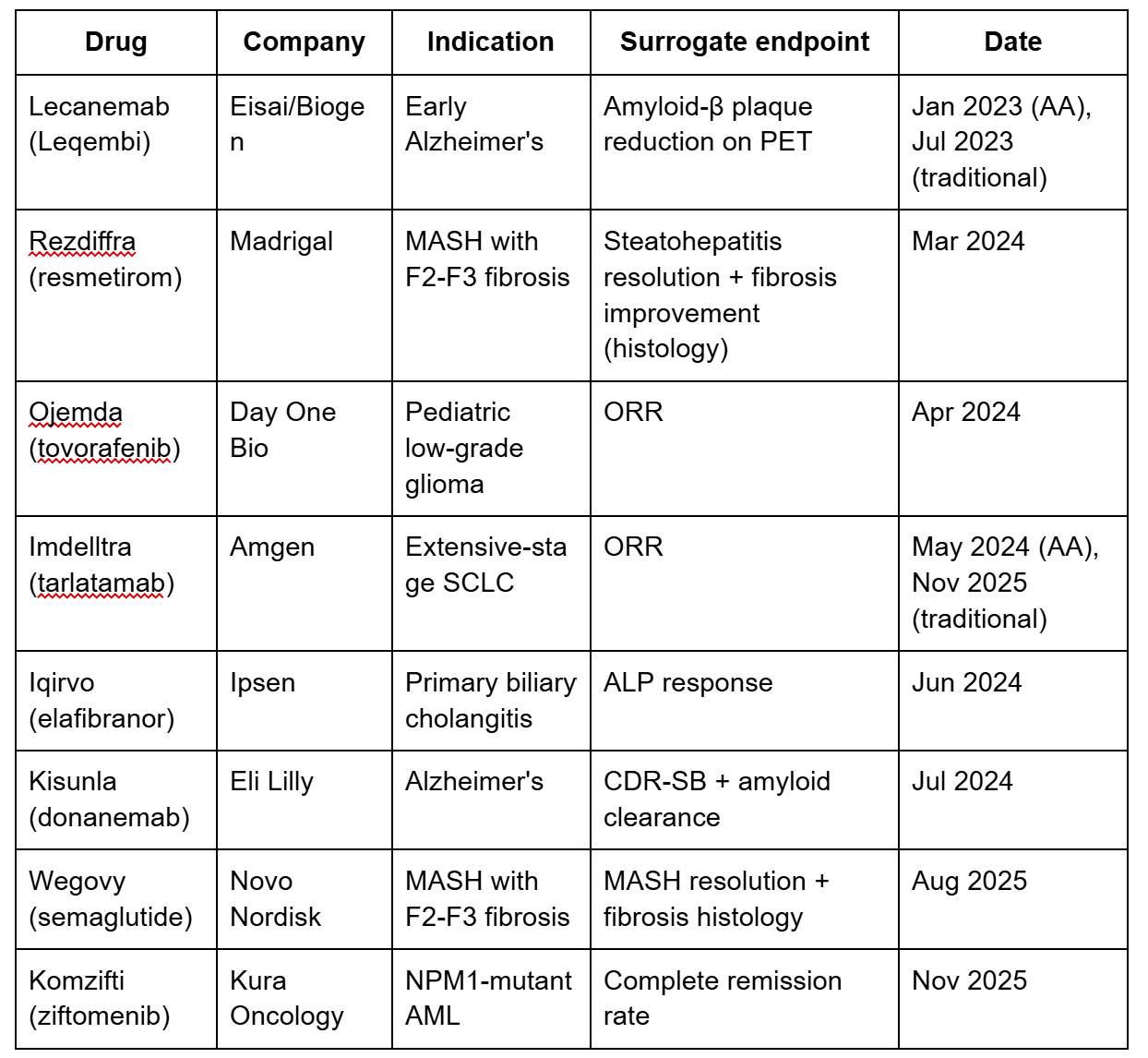
Surrogate endpoints enabling accelerated approval (2023-2026)
Biomarker absence shapes entire development strategies
MASH: the $100B endpoint problem. For decades, the only gold standard for MASH was liver biopsy, which is invasive, sampling only 1/50,000th of the liver, with inter-reader kappa scores of just 0.33-0.61. Screening failure rates are astronomical (~13% of biopsied patients don’t confirm MASH). This forced Phase 3 trials like Madrigal’s MAESTRO-NASH to require serial biopsies (expensive, slow, and limiting enrollment). The paradigm shift came in September 2025 when FDA accepted FibroScan/VCTE (vibration-controlled transient elastography) as a “reasonably likely surrogate endpoint”. It was the first non-invasive test accepted for MASH. Lilly immediately announced plans for Phase 3 trials of tirzepatide using purely non-invasive assessment. Additionally, FDA qualified AIM-MASH, the first AI tool for MASH histological scoring (Nature Medicine 2024), to reduce biopsy variability.
Example where biomarker strategy was inadequate
CETP inhibitors: the $10B+ surrogate biomarker trap. Torcetrapib (Pfizer), dalcetrapib (Roche), and evacetrapib (Lilly) all successfully raised HDL cholesterol—the surrogate that was “reasonably likely to predict clinical benefit.” Torcetrapib increased death risk. Dalcetrapib showed zero efficacy. Evacetrapib was stopped for futility. The surrogate moved perfectly; the clinical outcome never followed. This remains the canonical warning against anchoring development strategy on an inadequately validated surrogate.
3. Model selection
Strategic model choices that de-risked programs
Emulate / Liver-Chip (organ-on-chip for DILI prediction). Validated across 870 chips, Emulate’s Liver-Chip detects drug-induced liver injury with 87% sensitivity. Economic modeling estimates broad adoption could reduce small-molecule liver toxicity failures by up to 87% and generate $3 billion/year in R&D productivity gains. Now used by 21 of the top 25 pharma companies and submitted to FDA’s ISTAND program.
Biocytogen (drug-target humanized mice for IND toxicology). Rather than using non-human primates, Biocytogen developed mice with specific human gene replacements. Over 20 drug candidates supported by these models have received IND approvals from FDA, NMPA, and TGA. This aligns with FDA’s April 2025 announcement to reduce NHP reliance.
Vivodyne (robotic 3D organ tissues + AI). Founded by organ-chip pioneer Dan Huh’s group, Vivodyne’s robotic Data Engine tests 10,000+ independent human tissues per run. Secured $38M seed (2024) + $40M Series A (May 2025, Khosla Ventures lead). Already partnered with a majority of top-10 pharma companies. Their deliberate bet on throughput (100,000+ tissue tests in 2 weeks) addresses the scalability gap that historically limited organoid adoption.
Recent failures attributable to wrong model selection
Takeda / TAK-875 (fasiglifam, GPR40 agonist, T2D). Phase 3 terminated due to severe DILI despite clean preclinical animal safety profiles. The reactive acyl glucuronide metabolite caused covalent binding above the DILI risk threshold and inhibited bile acid transporters, the mechanisms standard rat and NHP assays failed to capture. Retrospective testing on Emulate’s Liver-Chip detected the toxicity signal.
Merck KGaA / Evobrutinib (BTK inhibitor, MS). Phase 3 halted for liver injury even though preclinical safety profile was clean. A 2025 Nature Communications ToxPredictor model trained on human hepatocyte RNA-seq correctly classified evobrutinib as “high risk” with low margin of safety. Standard preclinical species missed it.
Alzheimer’s disease mouse models. Over 155 terminated AD clinical trials as of 2022. A 2024 eLife paper documented that transgenic AD mice (5XFAD, 3xTg-AD, APP/PS1) express familial AD mutations found in <1% of all AD cases. Past amyloid vaccines worked in mice but caused brain swelling in humans. Second-generation humanized App models and iPSC-derived cerebral organoids are now emerging as alternatives.
The model decision framework
The most sophisticated programs now build tiered decision trees: (1) 2D cultures for initial high-throughput screening, (2) organoids/organ-chips for human-specific efficacy and toxicity questions, (3) humanized mice for in vivo target engagement and systemic PK/PD, (4) PDX models for tumor heterogeneity, (5) standard animals for regulatory-required systemic toxicity.
The regulatory landscape supports this: FDA Modernization Act 2.0 (2022) removed mandatory animal testing. The FDA’s April 2025 roadmap aims to make animal testing “the exception rather than the norm” within 3-5 years, starting with monoclonal antibodies. The PDX model market is valued at ~$540M in 2025, projected to reach $1B by 2030.
4. Modality constraints
Target location as the primary modality determinant
Extracellular targets → antibodies. AbbVie acquired Aliada Therapeutics for $1.4 billion (October 2024) for its anti-pyroglutamate amyloid-β antibody with BBB-crossing technology (MODEL™ platform using transferrin receptor and CD98 binders). AbbVie had already killed its undifferentiated internal anti-amyloid antibody ABBV-916. The mAb clinical pipeline is the largest across all modalities, with 18% growth in the past year.
Intracellular targets → alternative modalities. Tecelra (afamitresgene autoleucel), approved 2024 for synovial sarcoma, is a TCR-T therapy targeting intracellular MAGE-A4 presented on cell surfaces via HLA. The target is inside the cancer cell and only accessible via MHC presentation, antibodies cannot reach it. This is also why targeted protein degraders (PROTACs, molecular glues) are gaining traction: ~85% of the human proteome remains “undruggable” by conventional small molecules, and these targets are overwhelmingly intracellular.
Gene/RNA targets → editing and silencing.
Verve Therapeutics / VERVE-102 (PCSK9 base editing): Single IV infusion achieved 53% LDL-C reduction and 60% PCSK9 reduction at highest dose. Eli Lilly acquired Verve in June 2025. The rationale: PCSK9 mAbs have ~50% discontinuation rates; inclisiran (siRNA) has ~20% first-year discontinuation. One-time permanent gene inactivation addresses the adherence crisis.
CRISPR Therapeutics / CTX310 (ANGPTL3 gene editing): Single IV dose achieved -73% ANGPTL3, -55% triglycerides, -49% LDL (published NEJM November 2025). Compared to Regeneron’s evinacumab (anti-ANGPTL3 mAb requiring monthly IV infusions), gene editing offers a one-time cure.
Intellia / NTLA-2001 (TTR gene editing): >90% TTR knockdown sustained over 2+ years, now in two Phase 3 trials.
Delivery as the rate-limiting step (and value driver)
Three data points frame the delivery challenge: more than 98% of small molecules and nearly 100% of large molecules fail to cross the blood-brain barrier at therapeutic concentrations. All six FDA-approved siRNA therapies target liver-expressed genes via GalNAc conjugation → Valuable billion-dollar assets in cracking extrahepatic delivery.
The deals confirm this thesis:
Novartis / Avidity Biosciences ($12 billion acquisition, October 2025): Avidity’s antibody-oligonucleotide conjugates (AOCs) fuse anti-TfR1 antibodies with oligonucleotides to deliver RNA therapeutics directly to skeletal and cardiac muscle. Lead program del-zota achieved ~25% normal dystrophin restoration in DMD (versus ~1% for eteplirsen’s naked PMO). Same payload, radically different delivery, 25x the efficacy.
Eli Lilly / Sangamo ($1.4 billion milestone deal): Licensed Sangamo’s STAC-BBB capsid (a proprietary AAV that crosses the blood-brain barrier) for CNS gene therapies.
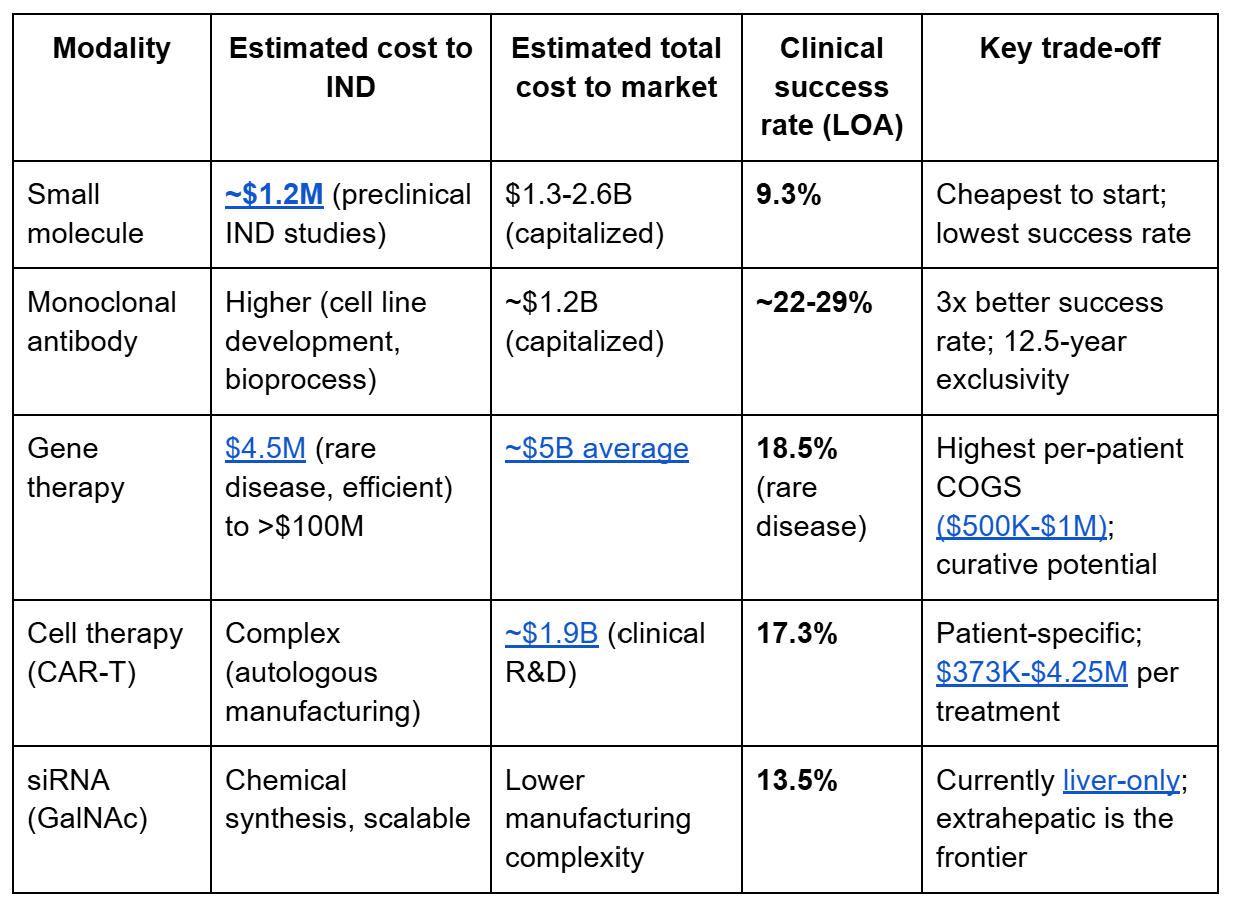
Cost and timeline hierarchy across modalities
IP forces modality diversification
The crowded PCSK9 antibody patent landscape (Amgen/Regeneron composition patents) likely incentivized Verve to pursue base editing and others to develop oral small molecules. ADC patent filings have more than doubled since 2018, reaching ~500 new patent families per year with 3,761 families analyzed. Biologics enjoy 12.5 years of market exclusivity versus 5 years for small molecules. It’s a powerful incentive to choose biologic modalities when the target biology permits it.
5. Clinical endpoint strategy
A clinical endpoint is a precisely defined variable intended to reflect an outcome of interest that is statistically analyzed to address a specific research question. The definition typically specifies the type of assessment made, the timing of those assessments, the tools used, and how multiple assessments within an individual are combined. Endpoints are not inherent properties of diseases, they are constructed measures that the FDA and your trial are designed around.
The FDA distinguishes three families of endpoints:
Primary endpoints are the basis for determining whether the trial meets its objective. They are the centerpiece of the regulatory submission and the trial must be statistically powered to detect a treatment effect on them.
Secondary endpoints provide supportive evidence about the drug’s effect on the primary endpoint or demonstrate additional clinical effects. They are analyzed only after the primary endpoint achieves statistical significance (per FDA’s hierarchical testing framework), and they can support additional labeling claims. But secondary endpoint results cannot save a missed primary endpoint.
Exploratory endpoints generate hypotheses for future trials. They have no regulatory weight but can inform dose selection, identify responsive subpopulations, or reveal unexpected effects.
Within these families, endpoints fall into two fundamental categories. Clinical outcome assessments directly measure how a patient feels, functions, or survives, such as pain scores, cognitive scales, overall survival, or physical function tests. These measure the thing we actually care about. Surrogate endpoints are biomarker-based substitutes that predict clinical benefit without directly measuring it, like tumor shrinkage predicting longer survival, or HbA1c reduction predicting fewer diabetic complications.
This distinction matters for regulatory strategy because it determines your approval pathway.
Co-primary and composite endpoints
When a disease affects patients through multiple dimensions, the FDA may require effects on more than one endpoint. Migraine trials use co-primary endpoints: pain freedom at 2 hours AND relief of the most bothersome symptom. Both must be met. This raises the statistical bar (multiplicity adjustments are required) but ensures the drug addresses the full clinical picture.
Composite endpoints combine multiple events into a single measure. MACE (major adverse cardiovascular events: cardiovascular death, non-fatal MI, non-fatal stroke) is the standard composite for cardiovascular outcome trials. The advantage is statistical efficiency: events accumulate faster when multiple outcomes count. The risk is that the composite can be driven by a less important component. A drug might reduce hospitalizations but not death, yet “meet” the composite endpoint.
Patient stratification that improved success rates
Donanemab‘s TRAILBLAZER-ALZ 2 stratified patients by tau burden on PET imaging. In the low/medium tau population, donanemab showed 35% slowing of CDR-SB decline versus placebo, a much stronger signal than in the overall population. Tissue-agnostic approvals (pembrolizumab for MSI-H/dMMR tumors; larotrectinib for NTRK fusions) represent the ultimate biomarker-enrichment strategy: single-arm basket trials enriched entirely by molecular biomarker, achieving high enough ORRs that randomized controls became unnecessary.
Biomarker-driven trials achieve 15.9% LOA, double the overall 7.9% average. A Boehringer Ingelheim-supported 2023 preprint demonstrated that AI-based stratification into “slow” versus “fast” Alzheimer’s progressors could make enrichment trials >13% cheaper with improved success probability.
The comparator problem
Your endpoint strategy must account for the standard of care at the time your trial reads out (not when it was designed). Pfizer’s KEYLYNK-010 in mCRPC randomized patients against abiraterone/enzalutamide, but the CARD trial established cabazitaxel as the new standard during enrollment. The STAMPEDE trial solved this adaptively: when docetaxel proved better, it became the new control arm for subsequent comparisons → continuous relevance.
6. The numbers that tie it all together
Success rates: the denominator matters most
Overall probability of success from Phase 1 to approval is 7.9% (BIO, 2011-2020) and trending downward to 6.7% (Citeline, 2014-2023). Phase 2 remains the biggest bottleneck at 28-29% success. But these averages hide massive variation:
Hematology: 23.9% LOA (highest)
Rare disease gene therapy: 18.5% LOA
CAR-T: 17.3% LOA
Rare disease overall: 17.0% LOA
Biomarker-selected trials: 15.9% LOA (2x average)
Oncology: 5.3% LOA
Chronic high-prevalence disease: 5.9% LOA
Cost benchmarks for founders
Average drug development cost reached $2.23 billion per asset in 2024 (Deloitte), up from $2.12B in 2023. Mean out-of-pocket cost per successful drug is $172.7 million (JAMA Network Open, 2024), rising to $515.8M when failure costs are included, and $879.3M when capital costs are added. Phase-level averages: Phase 1 ~$5.3M, Phase 2 ~$18.5M, Phase 3 ~$52.8M. But these mask modality-specific realities: rare disease gene therapy can go from discovery to NDA for under $25M (Elpida Therapeutics), while large-indication cardiovascular outcomes trials consume hundreds of millions.
The AI prediction frontier
A scoping review identified 142 studies (2013-2024) applying AI to clinical trial risk prediction, achieving up to 85% accuracy in forecasting outcomes and AUROC up to 96% for specific predictions. AI-accelerated trial design may reduce timelines by 30-50% and costs by up to 40%. The top predictive factors: disease indication, target, modality, drug novelty, and sponsor type.
Connecting the pieces: target typology as the organizing principle
The five decisions above are not independent. They form an interlocking system where the target typology determines the natural constraints and the founder’s job is to build a program architecture that is internally consistent.
A genetically validated extracellular target (Tier 2 evidence, clear LOF phenotype, accessible to mAbs) with a symptomatic endpoint is the lowest-risk, fastest-to-conviction profile. You can move quickly through validation, pick an established modality, use target engagement biomarkers to set dose, and power a pivotal trial on a clinical endpoint with weeks-to-months readout. Your primary risk is competitive as someone else is probably working on the same target. Your capital efficiency should be high.
A perturbation-derived intracellular target (Tier 3-4 evidence, requires novel modality, disease-modifying intent in a progressive condition) is the highest-risk, highest-reward profile. You need extensive orthogonal validation before IND, must solve a delivery problem, may lack intermediate biomarkers, and face a years-long timeline to clinical conviction. Your primary risk is biological, you might have the wrong target. Your capital requirements will be an order of magnitude higher.
Most programs fall somewhere between these extremes, and the strategic art is in honestly assessing where your target sits and building a plan that neither overreaches (spending as if you have Tier 2 evidence when you have Tier 4) nor under-invests (running a minimal preclinical package when your biology demands more).
This series has tried to lay out the logic of that structure, from how targets are identified and validated, to how development strategy follows from target biology. The gap between a scientific insight about a target and an approved medicine is technical, strategic, financial, and organizational. The founders who close that gap most efficiently are the ones who understand that every decision downstream (the biomarker, the model, the modality, the endpoint, the financing) is already constrained by the biology they chose at the start.
Build the plan that matches your biology. Then execute it faster and better than anyone else.
Acknowledgements
And that is the end of the “Betting on Biology” series! Appreciate everyone who has read and commented, as always suggestions of topics are welcome. A big thank you to Satvik Dasariraju for the inspiration, thoughtful comments, and prompt answers to my many questions; to Alex Colville for invaluable writing guidance throughout the process; and to every age1 crew for helpful pointers. Special thanks to Aerska, Loyal, and the amazing founders for the inspiration.
Cheers, see you next piece/series!
 | A guest post by
|