

The Cheapest Insurance in Drug Development
A framework for target validation before the million-dollar commitment


This week, Loyal raised a $100 million Series C to advance the first FDA-approved canine longevity drug: a caloric restriction mimetic targeting age-related metabolic dysfunction in senior dogs. Three days earlier, Aerska raised a $39 million Series A to deliver siRNA across the blood-brain barrier for genetic forms of Alzheimer’s and Huntington’s disease. These are very different companies building very different medicines, but both of them made rigorous efforts to validate their targets. Loyal’s lead program LOY-002 rests on decades of cross-species caloric restriction data and the largest veterinary clinical trial in history. Aerska’s platform targets genes with among the strongest genetic evidence in neurodegeneration, and solves the tissue-of-action problem that has killed prior CNS programs.
Identifying a plausible target is only the beginning. Target validation is the systematic process of confirming that modulating a specific molecular target will produce a therapeutic effect in human patients before committing to expensive clinical development. It is a structured body of evidence addressing a chain of linked questions: Is this target causally involved in the disease, or correlated with it? If causal, in which direction should it be modulated (inhibited or activated)? In which tissue must the drug act? In which patients, at which disease stage? And, importantly: what experiment could disprove all of this most efficiently?
Echoing the previous parts of the series: focusing on a wrong target is EXPENSIVE. 40%-50% of clinical drug development failures are due to a lack of clinical efficacy. Phase III trials, where most of this value is destroyed, have a median pivotal cost of $19M with large outcome trials routinely costing $100-350M. Including the cost of all prior failed programs, the fully-loaded cost per approved drug now ranges from $1.5 billion to $2.8 billion. Beyond financial loss, more importantly, a Phase III failure burns a decade of scientific effort and leaves patients without the therapy they were promised.
The data on what reduces this failure rate is unambiguous. Nelson et al.’s landmark 2015 Nature Genetics analysis showed that drug programs targeting proteins with human genetic evidence of disease association were twice as likely to gain approval. A 2024 follow-up in Nature that leverages a decade of additional GWAS data and pipeline outcomes revised that estimate upward: genetically supported targets are now 2.6× more likely to succeed, and the advantage increases with confidence in the causal gene. Alnylam Pharmaceuticals, which has made genetic target validation a foundational R&D principle, reports a clinical trial success rate 6× the industry average. And when AstraZeneca implemented its “5R framework“ (right target, right tissue, right safety, right patient, right commercial potential) its success rate from candidate nomination to Phase III completion rose from 4% to 19% in five years, and has since climbed to 23%, nearly six times the pre-reform baseline and well above the industry average.
This post lays out a framework for evaluating target validation when building a drug program. We cover directionality testing, context dependence, the hierarchy of evidence, and what we call “killer experiments”: the cheapest insurance money can buy.
Directionality testing: the billion dollar coin flip
A fundamental early question in target validation is the direction of intervention - should we activate (agonize) or inhibit the target? This decision must align with the disease biology. If the disease state already suppresses the target’s activity or levels, further inhibition may yield little benefit. For example, trials of myostatin inhibitors in Duchenne muscular dystrophy (DMD) failed partly because DMD patients already have very low myostatin levels. In such a context, trying to inhibit an already diminished factor is a losing battle. Conversely, if considering an inhibitory drug for a condition like obesity, one would prefer the target to be overactive or elevated in obesity – inhibiting something that is already low or normal in obese patients is unlikely to help. The target’s expression across age and disease states provides clues: ideally, an obesity target intended for blockade should be increased in obese individuals (and in contrast a target to agonize might be abnormally low or underactive in disease).
The GIP receptor directionality paradox
Glucose-dependent insulinotropic polypeptide (GIP) exemplifies how tricky directionality can be. Human genetics indicate that reducing GIP signaling might be beneficial: loss-of-function variants in the GIP receptor (GIPR) are associated with lower BMI and protection against obesity. This suggests that antagonizing GIPR could have anti-obesity effects, and indeed GIPR antagonists are being explored for weight loss, with Amgen’s MariTide achieving approximately 20% weight loss at 52 weeks.
Paradoxically, agonizing the GIP receptor has also shown promise when combined with GLP-1 agonism. The dual GIP/GLP-1 agonist tirzepatide produces approximately 21% weight loss in the SURMOUNT trials. How could activating a receptor that genetics say is better off silent lead to weight reduction?
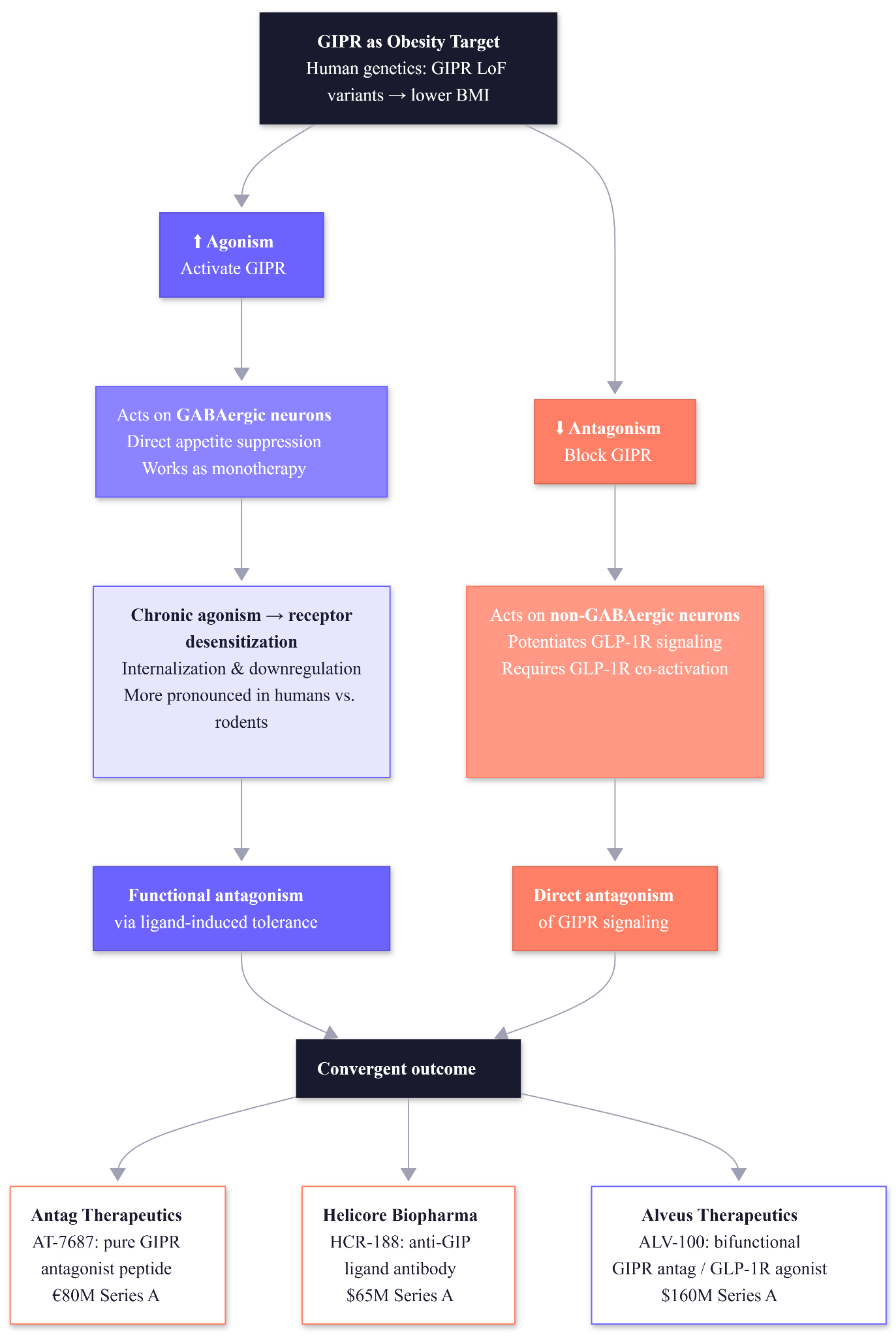
The resolution involves three mechanisms. Firstly, GIPR agonism and antagonism reduce food intake through different neuronal populations. Agonism acts on GABAergic neurons to suppress appetite directly, while antagonism acts on non-GABAergic neurons to potentiate GLP-1 receptor signaling. The practical implication is that antagonism requires concurrent GLP-1R activation to work, while agonism does not. Secondly, chronic agonism can lead to functional antagonism through receptor desensitization. Continuous stimulation of GIPR causes the receptor to internalize or downregulate, “burning out” the receptor’s activity. Thirdly, this effect seems more pronounced in humans than in rodents. Thus, a long-acting agonist can paradoxically achieve the same outcome as an antagonist by inducing ligand-induced tolerance.
The paradox and the mechanistic insights has spawned over $300 million in startup financing in the past fourteen months:
Antag Therapeutics (Copenhagen, €80M Series A, Versant/Novo Holdings) launched with AT-7687, a pure GIPR antagonist peptide co-developed by GLP-1 co-discoverer Jens Juul Holst. Phase 1 data reported in January 2026 showed enhanced weight loss when combined with the amylin analog cagrilintide — superior to either monotherapy. Preclinical data showed subnanomolar antagonistic potency (pKB 9.5) with a 27-hour half-life.
Helicore Biopharma (San Francisco, $65M Series A, Versant/OrbiMed) takes a different approach: its lead asset HCR-188 is a monoclonal antibody that binds circulating GIP ligand rather than blocking the receptor directly. This could neutralize GIP signaling in both periphery and CNS. Phase 1 is underway.
Alveus Therapeutics (Philadelphia, $160M Series A, New Rhein/Sanofi Capital) is advancing ALV-100, a bifunctional GIPR antagonist/GLP-1R agonist fusion. Phase 1b dosed its first patient in January 2026.
This shows the importance of testing both directions: a “directionality test” (e.g. comparing agonist vs antagonist molecules or genetic upregulation vs knockout) is critical to reveal which way of nudging the pathway yields benefit. It’s not always intuitive, human biology may respond differently than mouse models or simplistic assumptions. Rigorous experimentation is needed to confirm the correct strategy (for instance, testing whether sustained GIPR activation actually decreases signaling over time, and whether blocking GIPR outright has similar or distinct effects on metabolism). Such tests prevent pursuing the wrong approach on a valid target. In summary, getting the direction right can make or break a target: we must ask whether to floor the gas or hit the brakes, and the answer must be grounded in empirical evidence about the disease context.
Context Matters
Directionality is closely tied to when and where the target acts. Some targets produce opposite effects in different tissues or disease stages, which means that even a correctly identified target with the right directionality can fail if the biological context is wrong. We evaluate context across three dimensions: disease-state specificity, tissue of action, and cross-species conservation.
Disease-state specificity
GDF15, a hormone that suppresses appetite by acting on brainstem receptors (GFRAL), illustrates the problem. Pharmacologically raising GDF15 causes weight loss in preclinical models by reducing appetite. But in chronically obese individuals, GDF15 is often already elevated with no apparent effect on appetite or weight. Obese humans and rodents have significantly higher circulating GDF15 levels than lean controls, yet their food intake does not decrease accordingly, implying a form of resistance or desensitization. One hypothesis is that high GDF15 in obesity upregulates a negative regulator (the metalloprotease MMP14) that cleaves the GFRAL receptor, dampening downstream signaling. A “miracle” anorectic hormone in a short-term mouse test may fizzle in the chronic human condition due to feedback mechanisms.
The lesson: evaluating a target requires checking disease-specific context. Is the target pathway intact and actionable in the relevant tissue? Does the patient’s disease stage or concurrent treatments change the target’s baseline activity? For DMD, chronic corticosteroids and muscle degeneration changed myostatin’s baseline. For GDF15, long-term obesity changed receptor responsiveness. We must ensure the biology we aim to manipulate is not already maximally compensated by the body’s own feedback responses.
Tissue-of-action
Tissue-of-action determines everything downstream. This is the validation principle that shaped Aerska. Dozens of CNS programs have failed not because the target biology was wrong, but because the drug never reached the brain in sufficient concentrations. The blood-brain barrier excludes over 98% of small molecules and virtually all large biologics. An siRNA that silences target mRNA with exquisite specificity is worthless if it cannot cross that barrier.
Aerska’s brain shuttle conjugates siRNA payloads to transferrin receptor-targeting antibodies, exploiting the brain’s iron transport machinery to achieve uniform deep brain distribution. The competitive landscape validates this approach: AbbVie acquired brain shuttle company Aliada Therapeutics for $1.4 billion, and Regeneron/Alnylam signed a roughly $1 billion deal for CNS siRNA delivery. The tissue-of-action problem in CNS is being solved, which means genetic targets that were previously undruggable are becoming addressable.
BioAge Labs’ brain-penetrant NLRP3 inhibitor BGE-102 provides a complementary example. Phase 1 data showed 86% median reduction in hsCRP, substantially outperforming peripheral-only NLRP3 inhibitors. This suggests that central inflammation drives systemic effects more potently than peripheral inflammation alone. The tissue-of-action experiment asks “does the drug get there?” as well as “does getting there matter for the phenotype?”, and the answer to the second question tend to generate more value.
Cross-species conservation
A powerful and underappreciated form of context validation is cross-species conservation. If the same molecular signatures of a disease appear across species with shared environments and evolutionary history, the probability of causal relevance increases substantially. This is the core scientific insight behind Loyal’s approach.
Canine aging recapitulates human aging hallmarks with specific enrichments for telomere attrition (5.0×), genomic instability (2.5×), and loss of proteostasis (1.9×). Dogs and humans have cohabitated for ~23,000 years, share environmental exposures (diet, pollutants, stress), develop the same age-related diseases (cancer, cognitive decline, arthritis, cardiac disease), and show overlapping genetic determinants of lifespan: IGF1 pathway variants influence size and longevity in both species. A recent comprehensive review of canine immunosenescence confirms that dogs display the same age-related immune decline as humans: decreased naïve T-cells, inverted CD4:CD8 ratios, thymic involution, and elevated inflammatory cytokines (IL-1β, IL-6, TNF-α). Importantly, canine caloric restriction studies have already shown improved CD4:CD8 ratios and delayed thymic atrophy which directly mirror the mechanism LOY-002 is designed to exploit.
The limitations are real and worth stating clearly. Proteomic overlap between dog and human aging was weaker than transcriptomic overlap, likely due to species bias in the SomaScan assay (optimized for human proteins). The study was cross-sectional (different dogs at different ages, not the same dogs tracked longitudinally), single-breed (beagles), and relatively small (n=40). But as translational models go, the dog sits in a unique position: long-lived enough to develop genuine age-related pathology, genetically diverse enough to model human variation, and sharing the same environment as the humans it models.
Hierarchy of Evidence
Not all validation is created equal. In Part I, we described the hierarchy of target identification: human perturbational biology > human genetics > in vivo models > in vitro assays. A parallel hierarchy applies to validation evidence:
Tier 1: Orthogonal convergence in humans. Multiple independent methods of modulating the target in human systems produce the same outcome. This is the gold standard because it controls for off-target effects, model artifacts, and species differences simultaneously.
Tier 2: Human genetic validation with dose-response. Loss-of-function and gain-of-function variants in the target gene produce graded, directionally consistent phenotypes in human populations.
Tier 3: Cross-species conservation with mechanistic clarity. The same molecular signatures appear across species with shared evolutionary and environmental history, and the mechanistic pathway is well characterized.
Tier 4: Preclinical pharmacology alone. The target has been validated only in cell-based assays or animal models without human genetic support or cross-species mechanistic conservation.
Every step up this hierarchy substantially de-risks the program. Moving from Tier 4 to Tier 2 by incorporating human genetic evidence early is one of the highest-ROI activities in preclinical drug development. The hierarchy also matters for speed of iteration: Tier 1 and Tier 2 programs can fail fast on delivery or safety without wasting years debating target biology. Tier 4 programs often do not know they have the wrong target until Phase 2, by which point millions have been spent.
Three axis of target evaluation
When evaluating a prospective drug target, it’s useful to score it on three critical axes: causality, context, and controllability.
Causality: does perturbing the target change disease (in humans ideally)?
Context: which cell type, state, timepoint, comorbidity, ancestry?
Controllability: can we modulate it to the required degree, safely, in the right tissue?
In practice, these three axes are interdependent. The most compelling targets check all boxes: clear causality, relevant context, and feasible controllability. If one axis is lacking, the program is riskier. For example, a target might have great human genetic causality, but if it’s an undruggable protein, that’s a dead end (unless new tech emerges). Or a highly druggable target might fail if it turns out to be involved in too many normal functions (context/safety issue). By systematically assessing causality, context, and controllability, we ensure a holistic evaluation of target viability. A successful drug target must have a strong reason to believe it drives disease, act in a way we can intervene in the patient, and be amenable to pharmacological control. These principles guide us in choosing targets that are not only scientifically interesting but also actionable and translatable to therapy.
On Killer Experiments
Once a target looks promising on paper across causality, context, and controllability, the next step is a series of “killer experiments.” These are critical tests (often done in early research) explicitly designed to kill the project if the target isn’t truly valid. Instead of seeking only confirmatory evidence, we actively stress-test the target hypothesis. By setting up rigorous experiments that a false target would fail, we can save time and money by abandoning losers early.
We evaluate targets across nine categories of killer experiment. Each one tests a specific assumption that, if wrong, would invalidate the therapeutic program. Not every target requires all nine, but every target requires honest assessment of which experiments are most informative for its specific risk profile.
Directionality Test (Agonize vs. Inhibit)
Does modulating the target in your intended direction produce the expected phenotypic change in disease-relevant systems?
The GIPR paradox is instructive here. Test the effect of both activating and inhibiting the target. This determines which direction of modulation is therapeutic.
For aging targets specifically, directionality is complicated by the fact that many pathways have context-dependent effects (e.g., mTOR activation is beneficial during development and wound healing but detrimental during aging). The directionality test must specify the disease context
Domain-Specific Modulation
Does the effect depend on modulating a specific domain, isoform, or conformation of the target?
Probe different functional domains of the target with precise tools. Some proteins have multiple activities or domains (enzymatic site, scaffolding region, etc.). Using selective molecules or mutations that affect only one domain can reveal what aspect of the target is relevant to disease.
Scholar Rock’s apitegromab exemplifies this. By targeting only latent myostatin (avoiding GDF11 and activin A cross-reactivity), it met Phase 3 primary endpoints in SMA where broad myostatin inhibitors failed in DMD. In the Phase 2 EMBRAZE trial, apitegromab combined with tirzepatide preserved 54.9% of lean mass in 24 weeks during weight loss. The same target protein, but domain specificity determined success versus failure. This experiment is particularly relevant for complex targets with multiple binding partners or conformational states.
Tissue-of-Action Test
Does the target reside in the tissue your drug can reach, and is that tissue the site of disease pathology?
Confirm the target’s key site of action.
This is the experiment Aerska’s platform is designed to answer. BioAge Labs’ brain-penetrant NLRP3 inhibitor BGE-102 provides another example: Phase 1 data showed 86% median reduction in hsCRP which substantially outperformed peripheral-only NLRP3 inhibitors. This result suggests that central inflammation drives systemic effects more potently than peripheral inflammation alone. The tissue-of-action experiment asks “does the drug get there?” as well as “does getting there matter for the phenotype?” → the answer to which can generate more value!
Dose-Response & Reversibility
Does the phenotypic effect scale with the degree of target modulation, and is it reversible when modulation stops?
Demonstrate that the phenotype follows target modulation in a dose-dependent manner and can be reversed. A strong target should show a graded response: e.g. 50% inhibition yields partial improvement, full inhibition yields max improvement. This dose–response relationship supports causality (it’s a hallmark of true cause-effect). Additionally, if turning the target off produces a phenotype, then turning it back on (or letting it recover) should reverse the phenotype.
Orthogonal Perturbation
Do independent methods of modulating the same target produce convergent outcomes?
Use multiple independent methods to modulate the target and see if they converge on the same outcome. Check out the previous 3 blogs where we went deep in this for the typology.
Target Engagement → Phenotype Link
(”PK/PD chain”)
Can you trace a quantitative chain from drug exposure to target engagement to downstream biomarker to clinical endpoint?
Show that the degree of target engagement correlates with the degree of phenotypic effect. It tells us the minimum level of target modulation required for benefit.
Ibrutinib in CLL: covalent binding to BTK Cys-481 → >95% target occupancy at clinical doses → BCR signaling suppression (ERK, NF-κB) → lymphocytosis from nodal redistribution → 71% overall response rate. Resistance mutations at the covalent binding site (C481S) prove the drug works exclusively through this mechanism. Higher trough BTK occupancy correlates with improved progression-free survival across multiple BTK inhibitors (ibrutinib, zanubrutinib, acalabrutinib). This level of mechanistic completeness enables rational dose selection, predicts resistance mechanisms, and provides regulatory confidence.
Early Safety Liabilities Scan
Does the target have essential functions in healthy tissue that would create on-mechanism toxicity?
Assess potential safety issues before investing too far. Even in early validation, we can do “safety scans” in silico and in vitro: check if the target gene is expressed in vital tissues, examine human mutations, pathway analysis etc.
The activin pathway illustrates this. ACE-031’s broad activin receptor trapping caused epistaxis and telangiectasia from BMP9/10 inhibition in DMD trials: on-mechanism toxicity from hitting receptors in healthy vascular endothelium. The same vascular biology became sotatercept’s therapeutic mechanism in PAH only after the disease context was matched to the on-target pharmacology.
Disease State vs. Healthy Differences
*(Expression with Age/Disease)*
Is the target differentially expressed or active in the disease state versus healthy tissue?
Examine how the target (or its ligand) changes with age and disease progression.
For Pfizer’s ponsegromab study in cachexia, they enrolled only patients with GDF15 ≥1,500 pg/mL to ensure the disease was mechanistically driven by the target. In Loyal’s canine aging work, the omic data provides exactly this kind of disease-state characterization. It shows which pathways are differentially active in aged versus young dogs at the molecular level before designing the intervention.
Ligand vs. Receptor Dynamics
In a ligand-receptor system, is the bottleneck the ligand, the receptor, or the signaling machinery?
Test whether targeting the ligand or the receptor yields the desired effect, and whether acute vs. sustained stimulation differ. It tests how the system responds to continuous vs. pulsatile signaling and whether we should intervene at the level of the signal or the sensor.
When is enough validation enough?
A framework this comprehensive raises a natural question: how much validation is sufficient before proceeding to clinical development? The answer depends on the tier of evidence and the cost of being wrong.
For Tier 1 and Tier 2 targets with strong human genetic causality, the primary risks are delivery and safety, not target biology. These programs can afford to move faster on validation because the core hypothesis is supported by the strongest available evidence. The killer experiments that matter most are tissue-of-action (can the drug get there?), target engagement (does it hit the target sufficiently?), and early safety scans (will on-mechanism toxicity limit the therapeutic window?). Spending two additional years validating whether the target is causal is wasteful when human genetics has already answered that question.
For Tier 3 and Tier 4 targets, the calculus reverses. Here, the risk of having the wrong target entirely is significant, and the killer experiments around orthogonal perturbation, disease-state differences, and dose-response carry the most value. The investment in thorough validation before entering clinical development is the highest-ROI expenditure available, because the alternative is discovering the target was wrong in a $200 million Phase 2 trial.
The general principle: validate enough to know where the remaining risk sits, then design the clinical program to address that risk efficiently. A program with Tier 2 evidence and a clear tissue-of-action plan is ready for IND-enabling studies. A program with only Tier 4 evidence and untested directionality is not. The killer experiments table is not a checklist to complete exhaustively, it is more like a tool for identifying which assumptions carry the most risk for a given target and testing those assumptions first.
This is the fourth piece in the “Betting on Biology” series. In our last piece of the series next week, we’ll analyse the strategic decisions coming out of target identification and validation. Essentially, what should we do with this information? What factors should we consider and how are they influenced by the target discovery typology? See you soon.
Acknowledgements
A big thank you to Satvik Dasariraju for the inspiration, thoughtful comments, and prompt answers to my many questions; to Alex Colville for invaluable writing guidance throughout the process; and to every age1 crew for helpful pointers. Special thanks to Aerska, Loyal, and the amazing founders for the inspiration. Cheers!
 | A guest post by
|