

Longevity Primer 1: Epigenetic Clocks
A look at Horvath 2013



This piece is part 1 of a new weekly series diving into seminal papers in the aging space. If you’d like to be updated on when the next article will be released, follow us on Substack or Twitter.
Introduction
Epigenetic clocks fit strangely into longevity. For many, it’s the first introduction they have to the aging field - perhaps through stumbling across many of the companies offering kits to discover your “true biological age.” For those that want to dive deeper into what this actually means, it’s a painful first dose of how complicated aging science can be. To understand epigenetic clocks, you need at least a basic grasp of epigenetics, machine learning, and the biology of aging itself. Even worse, the idea of “understanding” epigenetic clocks is a false illusion - they are a composite of markers masquerading as one aging tracker. But we’ll get to that later.
On the flip side, as someone who has finished a PhD on the topic of epigenetic clocks, the discourse around them feels strangely unscientific. Ask anyone who has inhabited the aging space for longer than six months and they will have a very strong opinion on aging clocks, despite likely not having read a paper in the field. These takes are usually emotionally rooted; frequently out of a spirit of annoyance for how much attention they receive, or out of a paternalistic desire to protect the public from commercial actors. As a result, conversations regarding epigenetic clocks feel more like awkward political chats around the Thanksgiving dinner table rather than nuanced scientific discourse. Regardless, love them or hate them - epigenetic clocks are here to stay, and at the very least we can understand them a little better.
In this piece, I’ll dive into Steve Horvath’s 2013 DNA methylation age of human tissues and cell types, as it’s by far the most well-known study in the field. The study does not describe the first aging clock, nor even the first epigenetic clock, but it set the tone for every aging biomarker study that came after it. It tackles aging, cancer, monkeys, stem cells, epigenetics, mutations, and much, much more.
Alright, so what’s an epigenetic clock?
There are two concepts to understand here - epigenetics and machine learning.
Epigenetics is the layer of information the cell uses to figure out which DNA to turn into RNA. This is what separates a neuron from a skin cell despite them sharing the exact same genetic information. There are many “types” of epigenetics - from changing where genes are in the nucleus to altering the compactness of the genome. In the case of epigenetic clocks, the type we care about is a chemical change to the base pairs encoding the DNA, and very specifically to cytosine. This is what that looks like:

What does adding an extra carbon and three hydrogens to cytosine (referred to as “methylation”) do? It’s quite complicated and context-dependent, and well beyond the scope of this piece. But epigenetic clocks are built on a survey of hundreds of thousands of cytosine base pairs and their methylation state. Of these hundreds of thousands of base pairs (I’ll call them “sites” in the future), a few hundred are chosen to actually predict a person’s age.
How are these few hundred chosen? Using an algorithm known as “elastic net.” The elastic net algorithm is shown hundreds of thousands of potential methylation sites from a few thousand people and their ages (the training set) and then asked to predict age for a few thousand more (the test set). On one hand, it’s very simple and straightforward - it’s based on linear regression, meaning that the age prediction is literally just an addition of each site multiplied by the proportion of them that are methylated and some factor. See the below:

The algorithm seeks to find some balance between a small-ish number of sites to use for age prediction without compromising on accuracy.
On the other hand, how does the algorithm know which set of hundreds of sites to use for age prediction and which to not? It’s unclear and semi-random. If you remove the sites a model uses to learn age and then train a new model on the remaining sites, the model ends up performing just as accurately. You can do this more than once, too. If you’re curious about what biological information the clock is using to predict age, you have a very long and challenging road ahead of you - the actual function of each site is poorly (at best) understood. One technical detail that is often overlooked is that the clocks are not learning information from individual cells but rather from hundreds of thousands of cells consisting of potentially dozens of cell types - and the proportions of these cell types change with age and disease. It becomes pretty easy to see why epigenetic clocks become a headache as soon as you try to go beyond “predict this person’s age.”
So why are people so excited?
Epigenetic clocks are really, really good at predicting age. Let’s get into the actual meat of the paper:
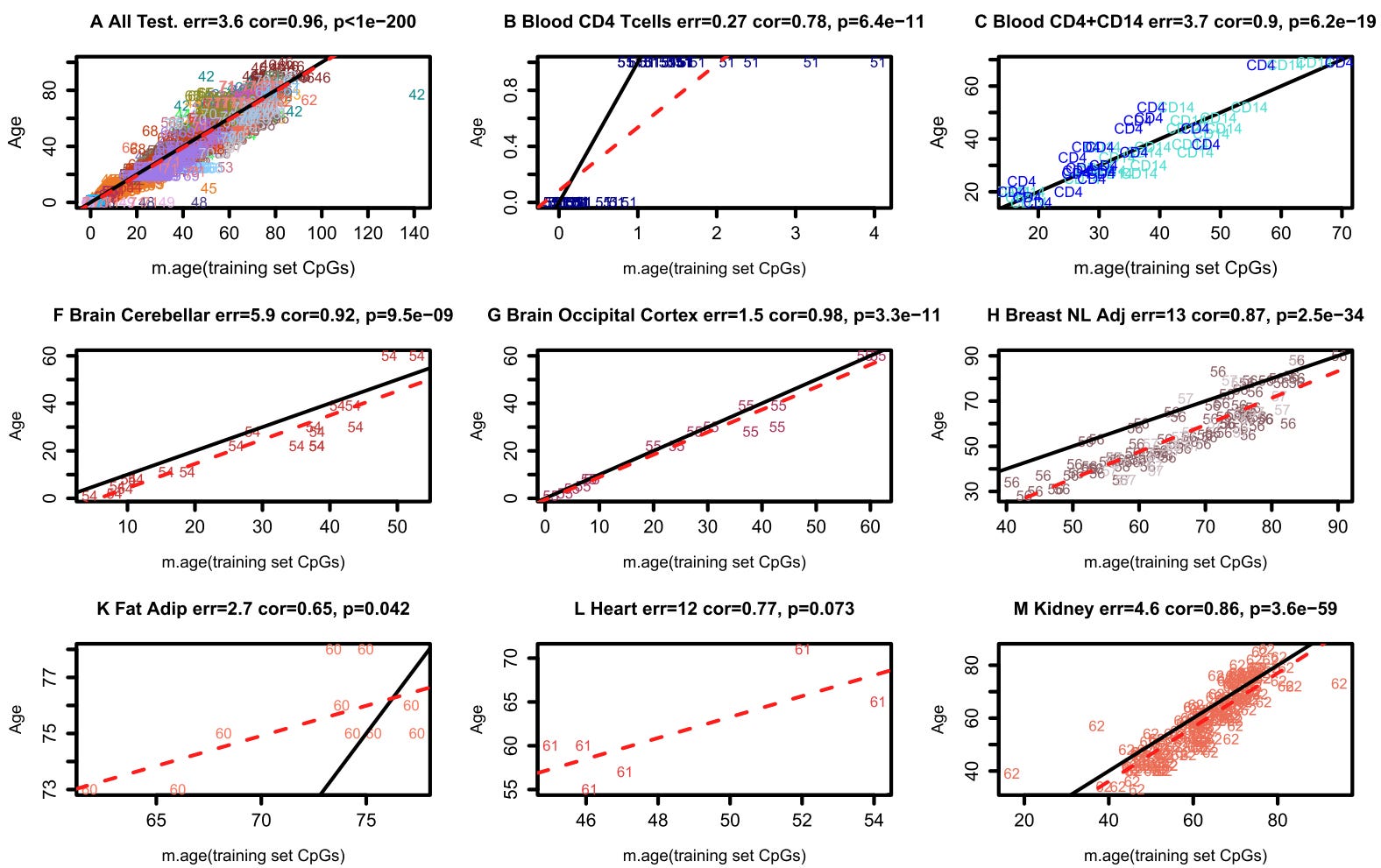
(A quick aside - the graphs in this paper are not aesthetically pleasing. This is one of the most cited papers in the longevity field but its figures are pretty rarely shown at conferences. I don’t want to editorialize too much here, but some of the coolest findings in this work are displayed in the ugliest figures, and it makes me wonder if some of the reason why the paper is mostly known for its clock development is because the aesthetic choices make the other findings very tough to display. It’s an incredibly thorough paper except when it comes to choices about how to show the data.)
In the figure above, the x axis represents an individual person’s predicted age and the y axis represents their chronological age. The error between them, on average, is 3.6 years. The Biomarkers of Aging Consortium is wrapping up an age prediction competition where the best models are clocking in at predicting age with an error of ~ 2.2 years. If you found a random person on the street and told them you could predict their age to within 2 years with a blood draw, they would be amazed. The fact that using DNA methylation - and so far, only DNA methylation comes close to this level of accuracy - gives you such a precise age estimate is fascinating. Even more interestingly, this age predictor works across a wide variety of ages, tissues, and species. Is there a case where the clock is not very good?
Surprisingly, yes. In sperm. Data below is from two different datasets, showing the predicted age of sperm (right bars) is well below the actual age of the donors (left bars).
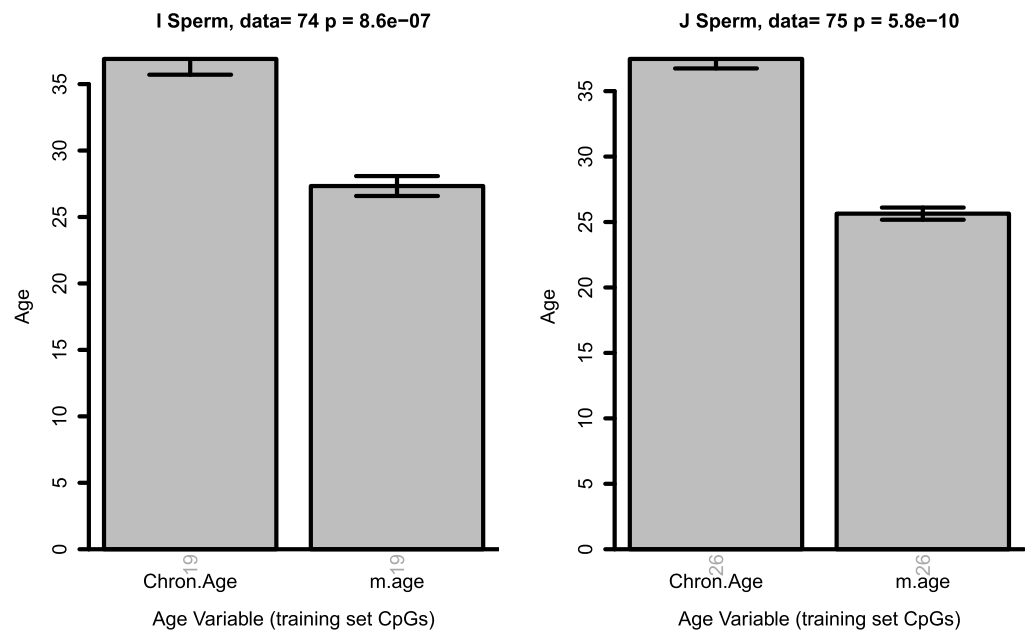
From figure 3
Why is the Horvath epigenetic clock - and in fact, every clock I’ve seen so far - so bad at predicting age in sperm? It’s not clear, and it’s a really interesting question to ask. Semen-specific epigenetic clocks have been developed, but they’re not nearly as good. They also rely on sites that seem to be immune-related, which makes me suspect they may be predicting age based on immune cells in semen as opposed to actual sperm cells. It adds support to an interesting line of discovery around looking for “rejuvenating” factors in germ cells, as they may age less relative to other cells (or, perhaps, not at all).
What other findings are there?
One extremely underrated finding from the paper is the pattern of change with age of the clock’s DNA methylation sites. I write about this in one of my other pieces, but almost every aging-related phenomenon has an exponential relationship with age. This is true especially of later manifestations of aging (i.e. disease) but also most molecular hallmarks. If you imagine aging as a feedforward mechanism that causes its own acceleration, this makes a lot of sense. But what about DNA methylation patterns used for age prediction?
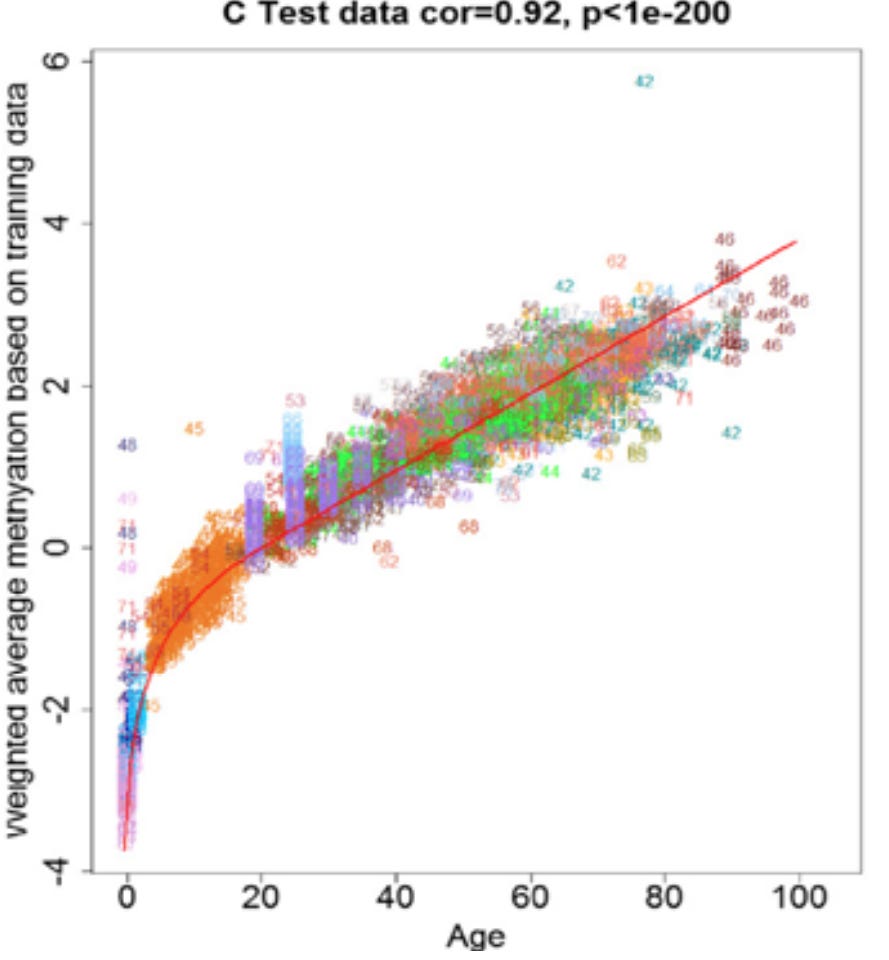
From figure 6
It’s a straight line up for development, and then a fairly linear relationship with age afterward. In my experience with epigenetic clocks, many predictive sites are used specifically to predict development (age < 16) and others post-development (age > 16), making them a composite measurement of development and aging. This is fascinating, and suggests that the DNA methylation patterns used for age prediction are not contributing to their own changes with aging. In other words, there is some other constant force that is changing DNA methylation patterns in a predictable manner.
“Ah, but you’re obviously going to find sites that have a linear relationship with age if you use a linear regression model to predict age!” you might say. That’s not the case - it turns out that DNA methylation at most sites changes linearly with age, especially after development. Epigenetic clocks have also been developed using non-linear models, and their predictive accuracies are not a whole lot better compared to what you would observe with a linear algorithm. This is a key finding from the epigenetic clock field, suggesting that epigenetic clocks are tracking something closer to the underlying cause of aging relative to other biomarkers. But this strays from the paper a little bit.
There are quite a few other interesting observations in this study, many of which contribute to our understanding of aging. A few of them suffer from having low sample sizes that make the resulting conclusions a bit hard to believe. Let’s take a few examples:

From figure 4
If you sample 5-7 chimpanzees or bonobos and then squint, it seems the Horvath clock can also predict age in other primates. It’s not terribly convincing here, but the line of thinking that we can use pan-species clocks has been reproduced in other studies and emphasized a previously-suspected link between development and aging.
In other areas, that’s not the case. One exciting avenue for studying aging is through understanding rare human diseases that look somewhat like accelerated aging (progerias). By surveying 3-4 patients with Werner’s syndrome (brown in figure below) and Hutchinson-Gilford Progeria (blue in figure below) syndrome, the study finds these diseases are not linked to accelerated epigenetic age:

From figure 2
Unsurprisingly, a later study with a higher sample size found that Werner’s syndrome patients do have an accelerated rate of epigenetic aging, though not to the extent that one might expect considering the progression of disease.
There are a few other neat findings. Embryonic stem cells have a very young epigenetic age, as do cells that are induced to be pluripotent stem cells:

From figure 5
If you then take these cells and let them divide a few dozen times, they start to have a higher epigenetic age. It’s pretty hard to know to what extent this reflects interesting biology as opposed to reflecting a technical artifact of having cells sitting in plastic. The data here have very high variability, making me somewhat lean towards the latter as a driver:

From figure 5
What about the cancer connection?
The paper makes a pretty big deal about cancer, which is a little strange considering the epigenetic changes associated with cancer would not a priori come to mind as being related to aging. In the main text, the case is made that epigenetic age prediction is tethered to genome instability, citing a negative relationship between age acceleration and number of mutations in cancer as evidence:

From figure 7. Each color is a unique dataset.
I have a hard time believing this line of argument considering how the data points become more spread out as the number of mutations increase. My interpretation here is that cancers with more mutations look less and less like normal human cells that the model was trained on, and thus their predicted ages become more unpredictable. Even this interpretation is a bit weak, as most of the data points from cancers with a small number of mutations come from one dataset which seems to drive most of the trend. The study tries to reinforce this trend by investigating a few cancer markers (FLT3, RAS, MLH1, BRAF) and checking their relationship with epigenetic age acceleration, but most of them end up not having much of a trend. The only convincing correlation is with estrogen receptor-positive vs. negative breast cancers:

From figure 8. Three other datasets show similar trends.
Maybe there’s something to this - the connection between estrogen receptor signaling and genome instability has been pretty well characterized.
Conclusions
The main takeaway to point out is that the paper does something that relatively few studies do in that it dedicates an entire section to theorizing on the mechanism of how epigenetic clocks work. I quote:

It’s a weird model because it suggests that embryonic stem cells do not try to maintain their epigenome at all, cells during development work very hard to maintain their epigenome, while adult cells have an unchanging level of epigenomic maintenance effort until death. Horvath then posits the following:
“If this EMS model of DNAm age is correct, then DNAm age should be accelerated by many perturbations that affect epigenetic stability. Further, age acceleration should have some beneficial effects given the protective role of the EMS. In particular, the EMS model of DNAm age entails the following testable predictions. First, cancer tissue should show signs of accelerated age, reflecting the protective actions of the EMS. Second, many mitogens, genomic aberrations, and oncogenes, which trigger the response of the EMS, should be associated with accelerated DNAm age. Third, high age acceleration of cancer tissue should be associated with fewer somatic mutations given the protective role of the EMS. Fourth, mutations in TP53 should be associated with a lower age acceleration of cancer tissue if one further assumes that p53 signaling helps trigger the EMS. All of these model predictions turn out to be true as will be shown in the following cancer applications.” Steve Horvath
Unfortunately, the evidence for these four model predictions ends up being pretty weak, and the underlying theory is rarely mentioned again. In 2018, the epigenetic maintenance system theory of epigenetic clocks ends up being replaced by something much more lukewarm by the same author:
“The proposed epigenetic clock theory of ageing views biological ageing as an unintended consequence of both developmental programmes and maintenance programmes, the molecular footprints of which give rise to DNAm age estimators.” Steve Horvath and Kenneth Raj
I bring this up not to be overly critical of the paper, but rather to draw a contrast between this high-level theorizing and the things the paper does really really well. The finding that DNA methylation clocks track a linear signature of aging - despite linear trends in aging being incredibly rare - is a huge finding that is rarely discussed. The observation that there are DNA methylation sites that track aging across tissues11 is fascinating, as is the fact there is even enough information embedded within the epigenome to allow for age prediction to within two years of chronological age. To me, the excitement here is that epigenetic clocks might be tracking something closer to the root causes of aging than any other biomarker found to date. The fact this is a cellular marker suggests that aging happens, first and foremost, in a cell-intrinsic manner before leading to systemic disruption.
There have been a few studies that have taken the best parts of this paper and really run with them. Early follow-ups by Brian Chen and many others investigated the difference between “intrinsic” (cellular) vs. “extrinsic” (cell composition-driven) aspects of epigenetic aging. Sylwia Kabacik’s 2022 paper (from the same lab) dives into how epigenetic age acceleration is linked to the molecular hallmarks of aging. Kejun Ying’s 2024 paper (Gladyshev lab) digs into which of the sites used for epigenetic clocks are causally linked to damage and adaptation. More and more papers are being released asking the right questions around what biological phenomena epigenetic clocks are tracking - from Meyer & Schumacher’s stochastic variation studies to Zane Koch’s investigation of somatic mutations.
But for every one paper using epigenetic clocks as a tool to study the fundamental nature of aging, there are ten prematurely hunting for associations with every clinical outcome in every possible study population. Without knowing what epigenetic clocks are even measuring, this is a case of putting the cart before the horse. And in this case, the fundamental aging biology horse may be much more interesting than the disease association cart.
A big thank you to Dr. Brian Chen, Alex Colville, Alex Kesin, Maggie Li, Zane Koch, Lada Nuzhna, Karl Pfleger, Aeowynn Coakley, and Ariel Floro for providing their thoughts and commentary in the writing of this piece. Stay tuned for next week’s longevity primer - we’ll be digging into partial reprogramming next!
It’s worth noting relatively few clock methylation sites actually track aging across most tissues - the model is mostly good at finding a combination of sites per tissue that are together universally predictive in every tissue.
 | A guest post by
|